Aberrant Hippocampal Neuroregenerative Plasticity in Schizophrenia: Reactive Neuroblastosis as a Possible Pathocellular Mechanism of Hallucination
bUniversity Grants Commission-Faculty Recharge Program (UGC-FRP), New Delhi-110002, India
Keywords
Abstract
Hallucination is a sensory perception that occurs in the absence of external stimuli during abnormal neurological disturbances and various mental diseases. Hallucination is recognized as a core psychotic symptom and is particularly more prevalent in individuals with schizophrenia. Strikingly, a significant number of subjects with Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD), and other neurological diseases like cerebral stroke and epileptic seizure also experience hallucination. While aberrant neurotransmission has been linked to the neuropathogenic events of schizophrenia, the precise cellular mechanism accounting for hallucinations remains obscure. Neurogenesis is a cellular process of producing new neurons from the neural stem cells (NSC)-derived neuroblasts in the brain that contribute to the regulation of pattern separation, mood, olfaction, learning, and memory in adulthood. Impaired neurogenesis in the hippocampus of the adult brain has been linked to stress, anxiety, depression, and dementia. Notably, many neurodegenerative disorders are characterized by the mitotic and functional activation of neuroblasts and cell cycle re-entry of mature neurons leading to a drastic alteration in neurogenic process, known as reactive neuroblastosis. Considering their neurophysiological properties, the abnormal integration of neuroblasts into the existing neural network or withdrawal of their connections can lead to abnormal synaptogenesis, and neurotransmission. Eventually, this would be expected to result in altered perception accounting for hallucination. Thus, this article emphasizes a hypothesis that aberrant neurogenic processes at the level of reactive neuroblastosis could be an underlying mechanism of hallucination in schizophrenia and other neurological diseases.
Introduction
Background of Schizophrenia
Schizophrenia is a debilitating neuropsychiatric disorder, characterized mainly by persistent hallucination, delusions, social withdrawal, emotional dysregulation, agitation, and cognitive deficits [1–3]. Initially, Emil Kraepelin described the clinical features of this peculiar affective disorder as dementia praecox, and manic depression due to the admixer of behavioral deformities overlapping with various mental illnesses [4]. Later on, the term schizophrenia was suggested by Eugen Bleuler in 1908 and he extended the description for the clinical signature of schizophrenia with the different positive and negative symptoms [2, 5]. While frequent episodes of hallucinations, delusions, paranoia, abnormal exhilaration, irrational thinking, and inexplicable behaviors are the positive symptoms of schizophrenia, the obvious negative symptoms comprise speech disorders, apathy, emotional blunting, and catatonia [6, 7]. The affective symptoms of schizophrenia include anxiety, depression, and suicidal thoughts, while an increased tendency of aggressive symptoms such as violent verbal abuse and assaultive behaviors are associated with schizophrenia [8, 9]. Besides, a considerable degree of memory loss, deterioration of interpersonal skills, and attention deficits are the key cognitive deficits noticed in schizophrenia [4, 10] (Fig 1).
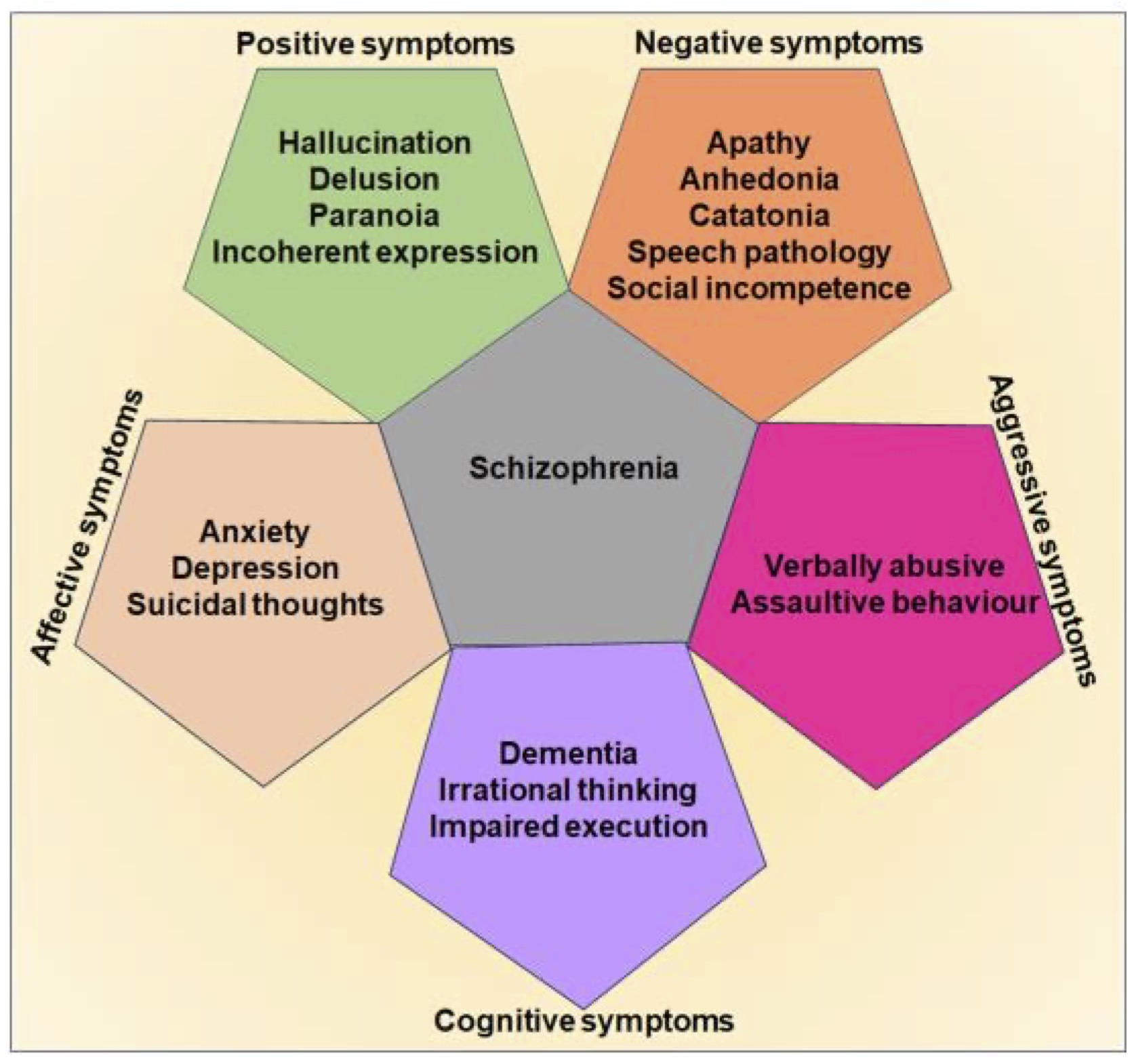
Fig. 1: Clinical symptoms of schizophrenia. The digital representation highlights the key positive, negative, affective, aggressive, and cognitive symptoms of schizophrenia.
The clinical symptoms of schizophrenia in childhood are rare, and the behavioral pathology typically develops in the late teenage [11, 12]. The prevalence of schizophrenia is almost 1% of the global population regardless of gender and ethnicity [1, 13]. Presently, there are no structured diagnostic strategies and confirmative biomarkers available to delineate the behavioral symptoms and pathogenesis of schizophrenia, because the etiology of the disease is not distinctive, the abnormal behavioral patterns are comorbid, vary among patients, and differ over time. The symptoms of schizophrenia appear to often co-occur with Bipolar disorder, Obsessive-compulsive disorder (OCD), and Major depressive disorder (MDD) [14, 15]. Despite the accumulation of enormous clinical reports and case studies, the precise causative factor and distinguished pathogenic mechanisms accountable for the onset and progression of schizophrenia remain to be established. There has been considerable progress made in the identification of therapeutic targets for schizophrenia with the aid of recent advancements in genome-wide studies, metabolomics, and proteomics approaches in combination with in silco platforms, genetic engineering tools, and stem cell technologies [16–18]. At present, tailored combinations of antipsychotics, antidepressants, and anxiolytic agents are widely used to manage schizophrenia [1, 19]. Though the neuropharmacological approaches, psychotherapy, and cognitive-behavioral interventions are useful in ameliorating the symptoms of schizophrenia, recurrence of clinical episodes has been reported in a significant percentage of patients regardless of the effectiveness of medicines and other types of therapeutic intervention [1, 20]. Considering the facts, schizophrenia patients require lifelong treatment. Moreover, the available medications appear to subsidize very few specific symptoms and the prolonged treatment has been associated with adverse changes in neurotransmitter levels, and metabolic, and hormonal profiles in schizophrenia patients [19, 21]. Therefore, there is a crucial need for scientific advancement in deciphering the underlying pathogenic determinants of schizophrenia which would help in identifying the ultimate therapeutic target.
Investigations into the underlying mechanisms of schizophrenia have taken many directions, with a particularly promising approach being the study of altered cellular populations in the brain. Focusing on the cellular perspective would provide crucial insights into the pathophysiology of schizophrenia, offering a deeper understanding of the biochemical, molecular, and cellular changes associated with the disorder. Reactive astrogliosis, a pathogenic cellular event associated with an overpopulation of astrocytes, has widely been regarded as a non-neuronal consequence leading to disruption in the electrochemical homeostasis of the brain and creating an imbalance in neurotransmitters during clinical episodes of various brain disorders [22]. Though experimental evidence highlights the alterations in the expression of genes related to astrocytes, the reports on the abnormal astrogliogenic events in schizophrenia are inconsistent [22–25]. Thus, the involvement of astrocytes in the pathogenic events in schizophrenia remains to be further established. Besides, a prominent histopathological signature for the activation of microglial cells, in part responsible for neuroinflammation has also increasingly been evident in the brains of schizophrenic subjects [26, 27]. The prolonged neuroinflammatory response has been known to impair the ongoing neurogenic process and synaptogenesis, collectively impairing the neuroplasticity in the brain [28–31]. Dysregulation of hippocampal neurogenesis is a prominent pathogenic characteristic of various neurodegenerative, mood, and psychiatric disorders including Alzheimer’s disease (AD), Huntington’s disease (HD), Parkinson’s disease (PD), stress, depression, anxiety, and schizophrenia [29, 32–38]. While the progressive decline in hippocampal neurogenesis contributes to the pathomechanisms of various forms of dementia in the aforementioned brain disorders, experimental evidence has established the occurrence of reactive neurogenic processes in the early phases of many neurodegenerative disorders [39–44]. In the physiological state, neuroblasts are considered immature neurons in the developing as well as the adult brain [45, 46]. The neurogenic process involves the proliferation of neural stem cells (NSC), their differentiation into neuroblasts, and the eventual maturation of these neuroblasts into fully functional neurons. The amount of neuroblasts produced in the neurogenic niches determines the degree of neurogenic process in the adult brain which is crucial for neuroregeneration and brain repair [45]. This ongoing neurogenesis plays a key role in the neuroplasticity of the hippocampus, contributing to cognitive functions and mood regulation. Recently, a new line of emerging scientific evidence has identified an abnormal activation profile of neuroblasts, termed reactive neuroblastosis [39, 44, 46–48]. This cellular process involves the overproduction of immature neurons leading to aberrant neurogenesis in the early phase of many neurodegenerative disorders. As neurodegenerative disorders progress into the later stages, the neuroblastosis events appear to be diminished due to the depletion or degeneration of the neuroblasts [44, 46, 47, 49]. Notably, traumatic brain injury, cerebral stroke, and epileptic seizure have also been characterized by reactive neuroblastosis and subsequent abnormal migration of neuroblasts in the affected regions of the brain [48, 50–52]. However, the role of reactive neuroblastosis in the pathogenic process and brain repair remains obscure. Remarkably, recent reports strongly indicate that subjects with AD, PD, HD, and other neurological diseases like cerebral stroke and epileptic seizure experience not only memory loss but also hallucination [53–55]. Considering abnormal neurogenesis as a common factor among both neurodegenerative and psychiatric disorders, the differential modulation of reactive neuroblasts along the disease course could indeed contribute to symptoms like hallucinations and dementia. As the ultimate cell fate of reactive neuroblasts is uncertain, the possibility of their involvement in neuropathogenesis accounting for psychiatric problems and memory functions is very high [44, 49]. The fluctuation in the terminal neurogenic process resulting from reactive neuroblasts in the adult brain could alter the neuroplasticity responsible for perception, memory, behavior, and consciousness, thereby inducing various psychological stimuli. Therefore, it can be speculated that incidences of hallucination could be correlated with altered neurogenic processes at the level of neuroblast turnover in the brain of subjects with schizophrenia. In the late phase of the disease, depletion of neuroblasts accounting for reduced neurogenesis could be associated with the deterioration of memory function. Therefore, the main aim of the review work is to establish the concept that the differential modulation of reactive neuroblastosis could be an underlying cellular mechanism of hallucination and dementia in schizophrenia. This manuscript has been formulated based on a comprehensive literature search encompassing the clinical, neurophysiological, behavioral, genetic, cellular, and neurogenic aspects of schizophrenia and abnormal neurogenesis processes in other brain disorders that display hallucination and dementia. Various search engines including PubMed were utilized to identify reports on schizophrenia and the regulation of neurogenesis. Highly relevant and recent articles have been considered to prepare the descriptive part, interpretations, and proposed hypothesis in the manuscript.
Risk factors and etiopathological relevance of schizophrenia
The clinical manifestations of schizophrenia are believed to originate from multifactorial elements including some definitive and mostly sporadic gene mutations, copy number variations, epigenetic alterations, dysregulated transcriptomics, chromosomal aberrations, metabolic defects, abnormal brain development, synaptic dysfunctions, impaired neurotransmission, substandard lifestyle and environmental factors [2, 56]. Notably, several key risk factors for schizophrenia have been identified, including maternal malnutrition, preeclampsia, gestational diabetes, prenatal viral infections, vitamin D deficiency, twin gestation, emergency cesarean section, childbirth complications, birth during the winter season, low birth weight, autoimmune diseases, chronic mood disorders, asphyxia, air pollution, illiteracy, and substance abuse, immigration to the foreign country, transcultural influences, living in an urban area and unsuitable environment [2, 57–60]. These diverse factors contribute to the complex interplay of genetic, environmental, and developmental influences implicated in the onset and progression of schizophrenia. Understanding and addressing these risk factors are crucial for effective prevention and management strategies for the disorder. As the mechanisms underlying the pathogenic basis of schizophrenia remain undetermined, the neurodevelopmental hypothesis has widely been considered for the onset and progression of schizophrenia. This hypothesis suggests that disturbances in early brain development, possibly influenced by genetic and environmental factors, predispose individuals to the development of schizophrenia later in life [61, 62]. Understanding the role of early brain development in schizophrenia is crucial for elucidating its etiology and developing more effective therapeutic interventions. During embryogenesis, the generation of neuroblasts from embryonic stem cells (ESCs) is crucial for the development of the brain [63]. The abnormal in-utero condition affecting brain development at the level of neuroblast formation has been considered a prime risk factor for schizophrenia [64, 65]. Therefore, congenital neurogenic defects have been speculated as the underlying cause for the onset of psychotic symptoms and cognitive deficits in the adult stage of life in individuals with schizophrenia [43, 66]. The migration of neuroblasts plays a key role in the morphogenesis of the brain and contributes to the neuroplasticity of the adult brain [67, 68]. Few studies have suggested that the aberrant migration of neuroblasts during early development could be linked with the pathogenesis of schizophrenia in the late stages [65]. A recent study by Jing Yang Tee 2021 indicated that NSCs derived from schizophrenia patients show increased migratory potential than control [69]. Another study by Bon Seong Goo et al. 2023, revealed abnormal neuronal migration in mice and human organoids due to a deficiency in mitotic arrest deficient-1 like (MADL), a schizophrenia-associated gene that regulates the polarity of migrating neurons [70]. Moreover, abnormalities in the disrupted-in-schizophrenia 1 (DISC1) and neuregulin (NRG)-1, the candidate risk genes associated with schizophrenia have been linked to disruption of neuronal migration [65, 71, 72]. The consequences of a reduction or mutation in DISC1 on adult neurogenesis were investigated in experimental mice by Duan et al. Their research found that suppression of DISC1 transcription provoked accelerated dendrite development and migration of neuronal progenitor cells, leading to inappropriate positioning of new neurons in the hippocampus [73]. Therefore, the genetic defects related to neuronal migration in the experimental models of schizophrenia appear to be inconsistent. Moreover, reports on the abnormal migration of neuroblasts in the hippocampus of postmortem brains from individuals with schizophrenia are limited [74]. Thus, the concept linking neuronal migration with the pathogenesis of schizophrenia requires additional scientific confirmation in human studies.
Next, the genes responsible for signal transduction pathways that regulate cell cycle such as iingless-integrated (Wnt), extracellular signal-regulated kinase (ERK), and protein kinase B (PKB) have been reported to be differentially regulated in schizophrenia patients [75, 76]. Further, mRNA levels of cell cycle regulators such as cyclin-dependent kinase (CDK)-4, minichromosome maintenance complex component (MCM)-7, and DNA polymerase delta subunit (POLD)-4 were found to be downregulated in individuals with schizophrenia [77]. Taken together, analysing the gene expression profiles related to the cell cycle and neuronal migration in postmortem brain samples has become crucial for understanding the underlying mechanisms of schizophrenia.
Several other theories have also been postulated for the neuropathogenic basis of schizophrenia. Disruption of glutamate transmission in the thalamocortical areas has been linked to the development of schizophrenia. Various experimental evidence gathered from the use of anesthetic agents namely phencyclidine and ketamine suggest that defects in the expression and function of glutamate decarboxylase (GAD)-1, hypofunction glutamate, and N-methyl-D-aspartate (NMDA) receptors are associated with schizophrenia [78, 79]. While astroglial cells are involved in the neurotransmission of glutamate at the synapses, abnormal astrogenesis during brain development has also been proposed to contribute to the progression of schizophrenia [80]. Further, unusual flux in the release of dopamine and differential expression of its receptors in mesolimbic areas, nigrostriatal, and mesocortical tracts have also strongly been linked to the clinical symptoms of schizophrenia [81]. Increased release and hyper-transmission of dopamine in the subcortical area of the brain are known to contribute to positive symptoms such as hallucinations and delusions in schizophrenia. Convergingly, hypofunction of dopamine resulting from decreased expression or inactivation of dopamine receptors in the prefrontal cortex and caudate nucleus appears to be associated with the development of negative symptoms like anhedonia, lack of motivation, and speech disorders [81, 82]. Experimental studies established from the use of antipsychotic drugs that modulate the serotonergic and dopaminergic systems have revealed that impaired interaction between dopamine and serotonin could prime the abnormal neurochemical events accounting for schizophrenia [83–85]. Furthermore, recent evidence indicates the dysfunctions of GABAergic neurons in the cortex, altered levels of serotonin, and defects in the cholinergic system of the brain during the symptomatic phase of schizophrenia [19, 56, 86, 87]. Additionally, increased levels of norepinephrine have also been suggested to play a role in the pathophysiology of schizophrenia [88].
Considering its heritable nature, many genetic determinants and variants have been linked to the pathogenesis of schizophrenia [89]. First-degree relatives and offspring of individuals with schizophrenia have a considerable chance of manifesting neuropsychological disturbances [57, 89–91]. However, the genetic linkages and the mutations are not unique among schizophrenia patients. The clinical episodes of schizophrenia have been mapped to various polymorphisms or dysregulation of susceptibility genes such as 1) NRG-1, a key factor involved in brain development and vesicular transport of glutamate and epidermal growth factor (EGF) signaling, 2) dystrobrevin-binding protein (DTNBP)-1 which aids in glutamate release, 3) catecholamine O-methyl transferase (COMT), important for signal transduction of dopamine, 4) dopamine beta-hydroxylase (DBH) that catalyzes the hydroxylation of dopamine and some phenylethylamine derivatives, 5) regulator of g-protein signaling (RGS)-9, responsible for various molecular pathways transduction in the brain and 6) DISC-1, involved in downstream dopamine signaling pathway [92–97]. Eventually, the suicidal behaviors in schizophrenia have been attributed to defects in genes such as the corticotropin-releasing hormone receptor (CRHR)-1 and corticotropin-releasing hormone binding protein (CRHBP), which encode stress response elements involved in the regulation of the hypothalamic-pituitary-adrenal (HPA)-axis [98]. Notably, the aforementioned neurobiochemical and genetic determinants and risk factors appear to be associated with morphological differences and neuroanatomical abnormalities in schizophrenia.
Neuromorphological and pathological alterations in schizophrenia
Owing to its obvious abnormalities in neurotransmission, there has been overwhelming data available for the description of the behavioral deformities and psychotic symptoms in schizophrenia [99]. However, the distinct neuropathological changes of schizophrenia arising from various idiopathic factors have long been refractory to the diagnosis and confirmative tests. Earlier attempts using radiology-based pneumoencephalography unveiled dilated lateral and third ventricles in the brains of subjects with schizophrenia [100, 101]. Subsequently, Johnstone et al. confirmed the presence of enlarged ventricles in schizophrenia patients using axial brain scans [102]. Considerable scientific and technological advancements in recent decades have enabled the implementation of neuroimaging techniques, and neuromorphometric assessments to differentiate the cytoarchitectural alteration and identify the functional defects in the brains of subjects with schizophrenia and those at risk of developing symptoms. In the quest to elucidate the neuropathological changes in the brain, compelling neuroimaging evidence obtained from various medical scanning modalities including computed tomography (CT), magnetic resonance imaging (MRI) with diffusion-tensor imaging (DTI) tractography, magnetic resonance spectroscopy (MRS), magnetoencephalography (MEG) and positron emission tomography (PET)-based studies have collectively demonstrated and validated enlarged ventricles, grey matter loss, structural deformities and structural loss in the corpus callosum, increased volume in basal ganglia, loss of myelination and dysconnectivity of neural network, differences in neurite curvature in schizophrenia [101, 103–105].
To note, the enlarged ventricle has been established as a prominent neuropathological hallmark related to tissue remodelling processes in many neurocognitive diseases as the neuroblasts migrate from the subventricular zone (SVZ) to the pathogenic and degenerating brain sites [106, 107]. Initially, the increased volume of cerebrospinal fluid (CSF) has been proposed for the enlarged ventricles in schizophrenia [108]. The shrinkages in the cortical, striatal, and thalamic areas have been reported to result in ventricular enlargement in schizophrenia [109]. As the evidence for the occurrence of neurodegeneration in schizophrenia is highly limited, reduced grey area has been linked to a combination of neurodevelopmental, genetic, and environmental factors. Notably, reduced neurogenic processes and impaired migration of neuroblasts during the developmental stage have been proposed to contribute to the reduced grey area in the adult stage [65]. According to the neuropil hypothesis, drastic reductions in neural circuity, spine density, and dendritic volume are accountable for grey matter loss in schizophrenia [110]. The histopathological analysis of the postmortem brain samples from individuals with schizophrenia has revealed synaptic loss, rather than neurodegeneration can be responsible for the volume loss in many brain regions [111]. In line with the neurodegenerative hypothesis, few reports indicate the possibilities of pathological neuronal apoptosis and oxidative damage in schizophrenia. However, the role and rationality for the occurrence of apoptotic events in schizophrenia remain ambiguous. Recently, the cell cycle reactivation in mature neurons induced by neuroinflammatory cytokines has been reported to result in reactive neuroblastosis. The cell cycle re-entry of mature neurons would result in synaptic pruning or cell death, thereby contributing to reduced grey area in schizophrenia. As proof, some earlier studies highlighted the increased neuronal density in the key brain areas of schizophrenia patients [110, 112]. Eventually, demyelination resulting from the degeneration of oligodendrocytes has been predicted to be the reason for white matter lesions in the frontal cortex, hippocampus, and cerebellum in schizophrenia [84, 113]. A surface-based MRI analysis by Sprooten et al., 2013 intended the widespread cortical thinning, more predominantly in superior frontal, medial parietal, and lateral occipital regions during the early stages of schizophrenia [114]. Therefore, reports on reduced grey matter in schizophrenia require to be revisited and validated with functional neuroimaging and histopathological assessments.
Besides, an analytical study done by Sheffield et al., 2017 using resting-state functional magnetic resonance imaging (rs-fcMRI) revealed that the cognitive impairment noticed in schizophrenic individuals is associated with loss of functional connectivity within and between fronto-parietal lobe and cingulo-opercular networks [115]. Additionally, magnetic resonance spectroscopy and PET-based studies on the brains of schizophrenia patients reported that excitatory-inhibitory imbalance in the cerebello-thalamo-cortical and striato-thalamo-cortical loops, hyperfunction in the mesolimbic dopamine pathway and variations in dopaminergic content in the prefrontal cortex (PFC), anterior cingulate gyrus, and hippocampus contributes to the pathophysiological changes related to development of psychotic disorders in schizophrenia [116, 117] (Fig 2).
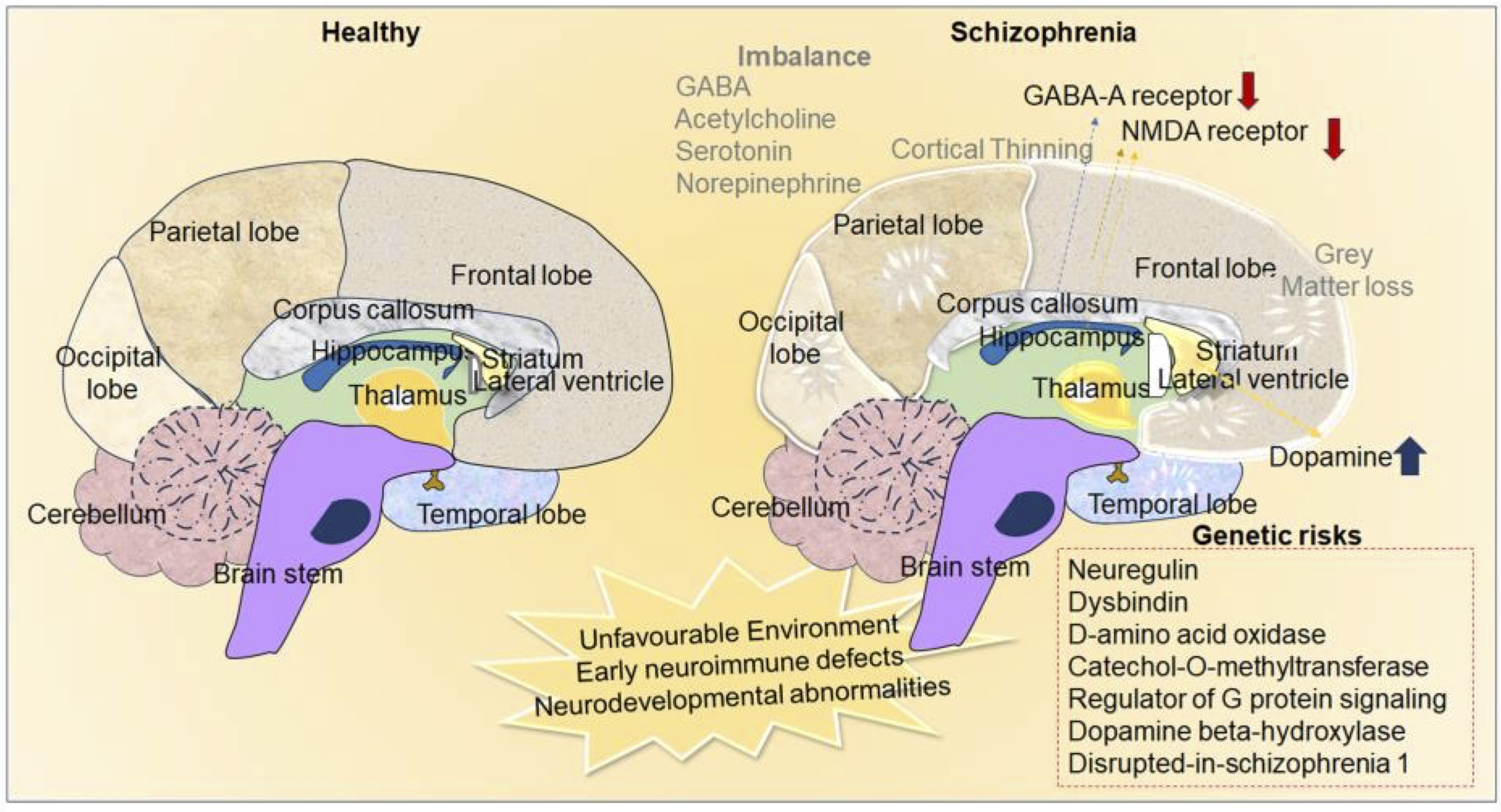
Fig. 2: Neurochemical and anatomical differences in the brain of healthy and schizophrenia. The image depicts the environmental impact, variations in neurotransmission, and hereditary factors as the etiological causes of schizophrenia. Neuroanatomical alterations such as cortical thinning and loss of grey matter in the schizophrenic brain compared to healthy brains.
A series of MRI-based reports on gross brain morphometric and gyrification assessments unveiled obvious volumetric reduction in the frontal and temporal lobes of schizophrenia subjects [76]. Concomitantly, decreased density of cortical regions and shrinkage in the amygdala, thalamus, nucleus accumbens, and hippocampus have been established as region-specific morphological defects in schizophrenia [120, 121]. Notably, the brains of subjects with schizophrenia have been characterized by reduced hippocampal volume [122]. The reduced hippocampal volume in schizophrenia has been attributed to several factors, including the reduced density of pyramidal cells in the cornu ammonis (CA) regions and decreased neuropil in the molecular layer and hilar region [123]. Similar to the reduced somal sizes noticed in pyramidal neurons of cortical areas, the decreased hippocampal volume observed in schizophrenia could be related to decreased somal sizes of pyramidal neurons in CA regions [124]. While some studies have reported reduced overall hippocampal density, few reports have indicated deformity in subareas such as CA1 contributing to the volume loss of the hippocampus [125]. In general, neuronal loss has been considered a primary reason for hippocampal atrophy in many brain and mental disorders [126, 127]. Schizophrenia patients with low serum levels of brain-derived neurotrophic factor (BDNF) and expression of its genetic variant BDNFMet have been characterized by a reduced hippocampal volume [128–130]. Defects in the BDNF signaling pathway have been proposed to result in neuronal shrinkage, and dendritic retraction in the diseased brain which might also contribute to the volume reduction in the hippocampus in many neurodegenerative disorders [131]. While few available postmortem studies of patients with schizophrenia revealed intensified BDNF signaling in the hippocampus, decreased levels of BDNF concentrations in the brain of schizophrenia were also evident [132–134]. However, subsequent studies revealed that there is no association between BDNF and hippocampus volume, therefore, the role of altered BDNF signaling on the hippocampal volume in schizophrenia requires additional investigation.
The extracellular matrix (ECM) contributes to a significant percentage of brain volume [135]. Studies related to abnormalities in reticular structure revealed marked differences in ECM components in the brains of subjects with schizophrenia [136]. Notably, the reduction of the perineuronal nets (PNNs), a key component of ECM that wraps around neurons, axons, and dendrites, gained scientific attention in schizophrenia research [137]. Reduced levels of PNNs have been correlated with structural disorganization and functional loss of key brain areas that are associated with motor sensory and cognitive functions [137, 138]. Thus, abnormalities in PNNs could also be a potential cause of hippocampal volume loss in schizophrenia regardless of neurodegeneration. In the meantime neurogenesis has been considered to play a role in structural and functional aspects of the hippocampus [97]. Therefore, the pathogenic role of ECM disintegration in association with abnormal hippocampal regenerative plasticity in schizophrenia could be an important aspect of scientific consideration.
Among various brain regions, the hippocampus has been considered to play a crucial role in neurocognitive functions as it holds a niche for NSCs [139, 140]. Atrophy or dysfunction of the hippocampus has been linked to dementia, mood, and psychotic disorders [29, 44, 46, 47, 139, 140]. Notably, schizophrenia has been characterized by neuroanatomical, cytoarchitectural, synaptic dissociation, demyelination, microglial activation, and functional abnormalities in association with neuroinflammation in the hippocampus [141, 142]. Distinctly, the ongoing neurogenesis mediated by NSCs-derived neuroblasts has been reported to play a key role in regenerative plasticity, memory, and mood functions [140]. In contrast, defects in the hippocampal neurogenic process have been linked to dementia, stress, anxiety, and depression-related symptoms in various neurological illnesses and traumatic brain injuries [29, 44, 47, 51, 52, 106, 123, 141]. Eventually, abnormal neurogenesis at the level of neuroblast turnover appears to be associated with neurodevelopmental disorders like autism [143]. Therefore, the possibilities for the involvement of aberrant neurogenesis in the hippocampus in the establishment of psychotic disorders cannot be excluded.
While the aging-related progressive cognitive decline has been correlated with a steady decline in hippocampal neurogenesis, recent evidence suggests varying degrees of neurogenesis upon the pathogenic progression in various forms of mental deterioration and neurodegenerative illnesses [144]. Notably, experimental models of schizophrenia have been characterized by arrest in the maturation of the hippocampus due to elevated levels of cellular and molecular signatures of immature neurons [145, 146]. As differential regulation, prolonged dysregulation, and impaired neurogenesis contribute to cognitive deficits, altered neural information processing generated by neurogenic processes in the brain might be associated with hallucinations and memory loss in schizophrenia. Thus, insight into mechanisms that overlap impaired neuroblastosis with hallucinations and dementia could provide advancement in understating the neurobiology of schizophrenia at the level of adult neurogenesis. However, the experimental evidence for the involvement of altered neurogenic processes with hallucination is highly limited in human situations. Indeed, many invitro and transgenic animal models of schizophrenia and the outcome of treatment with psychedelic drugs strongly point towards the possibilities for the potential link between altered neurogenesis and hallucination [117, 147–149]. Concurrently, individuals with various neurological diseases who experience hallucination have been found to exhibit abnormal adult neurogenesis [150, 151]. Therefore, the insight into the possible role of adult neurogenesis in the development of hallucination would offer complementary approaches to distinguish the shared pathocellular mechanisms among schizophrenia and other brain diseases.
Reactive neuroblastosis as an underlying mechanism of hallucination in schizophrenia and other neurological diseases
Hallucination is the involuntary emergence of illusory, perceptual, and mystical experiences of the brain that occur devoid of external stimuli through the sensory organs and environment [152]. Hallucination can occur at the level of auditory, visual, tactile, olfactory, and gustatory modalities during conscious as well as in paradoxical sleeping states. Functional neuroimaging studies suggest that the generation of abrupted neural impulses in the key brain areas including the sensory cortex, insula, putamen, and hippocampus could be an underlying cause of hallucination [153–155]. However, the origin of the spontaneous neural activity in the brain that synthesizes substrate for the generation of hallucination remains obscure. Thus far, numerous theories have been proposed for the underlying basis of hallucination [156, 157]. For example, Lopez Ibor proposed that abnormal activation of engram, a hypothetical form of substratum essential for the cognitive process in ideational centers of the brain, can be an underlying basis of hallucination [156, 158]. According to Hughlings Jackson, higher motor centers of the brain are pivotal for intellectual functioning when they are activated spontaneously or independently of peripheral stimuli. Conversely, the deactivation of upper motor centers can elicit the activation of mid-level motor centers, potentially resulting in episodes of hallucination [159, 160]. As per Wilder Penfield’s experimental findings, electrical stimulation in certain cortical or subcortical structures could induce different forms of hallucinations [161, 162]. Notably, the occurrence of hallucination is intricately linked to changes in neuroplasticity, particularly within key brain regions such as primary and secondary sensory cortices, basal ganglia, and limbic system including the hippocampus [163]. Among different neurotransmitter-based hypotheses, varying levels of dopamine in the limbic system have been strongly implicated in the development of hallucinations [164]. Emerging research evidence has also highlighted the involvement of other factors such as abnormal immune activation, increased cerebral blood flow, circulating metabolites, and energy metabolism in the brain in the manifestation of hallucinations [165–168]. However, most of the theories that explain hallucinations appear to be merely paradoxical and none have conclusively delineated the definite underlying mechanism of hallucination. Therefore, the pursuit of understanding the root cause of hallucination from various perspectives including at the cellular level has become increasingly important.
Though hallucination is a prominent psychotic symptom of schizophrenia, it also occurs in many other diseases and arises in response to some substance abuse [164]. Indeed, hallucination is a key clinical problem in diverged medical conditions including psychiatric complications and neurodegenerative diseases [169]. Various forms of dementia, bipolar disorder, OCD, epilepsy, cerebral stroke, migraine, brain trauma, brain lesions, brain tumors, metabolic disorders, and Charles bonnet syndrome have been known to be associated with a considerable degree of incidence with hallucination [170]. In addition, individuals with anxiety, stress, depression, and post-traumatic stress disorder (PTSD) have also been known to experience hallucination [171]. In AD, reduced acetylcholinesterase (AChE) activity in the brain has been predicted as a biochemical cause of hallucinations in a significant percentage of subjects with progressive memory loss [172, 173]. Eventually, PD and HD patients have also been reported to experience hallucinations which are attributed to imbalances in dopamine and GABA in the limbic system of the brain [55, 174]. However, the occurrence of hallucinations in neurodegenerative disorders is a subject of debate as many drugs that are used for the management therapy of neurological deficits, psychotic problems, sleeping abnormalities, and mood disorders have also been known to induce hallucinations [175]. While there are documented cases of hallucinations associated with prolonged intake of medications like antiepileptic drugs, antidepressants, anticancer medications, and sleeping pills such as narcotics, steroids, pentoxifylline, tramadol, bromocriptine, sertraline, and trazodone, it's important to note that these drugs also exhibit a notable impact on the regulation of hippocampal neurogenesis [176, 177]. Therefore, understanding a specific neuropathogenic signature of schizophrenia that overlaps with other neurological diseases and the mode of action of hallucinogenic drugs and their impact on the regulation of neurogenesis could provide a valid clue in understanding the underlying basis of the hallucinations.
During embryogenesis, the generation of neurons from embryonic NSCs-derived neuroblasts plays a key role in neurodevelopment, whereas defects in the early neurogenic process have been implicated in the pathogenic mechanisms underlying neurodevelopmental disorders and mental disturbances [178, 179]. Among various predictions, a potential link between schizophrenia and aberrant neurogenic events responsible for abnormal brain development in early life has been widely considered to have a negative impact in the latter adulthood stage leading to psychiatric disturbance and neurocognitive impairments [43, 180, 181]. Thus, there has been considerable scientific interest in exploring the alteration of neurogenesis in schizophrenia [182]. In the healthy brain, the degree of neuroblasts generation, accounting for the neuroregenerative characteristics has been directly correlated with mental health, intellectual ability, sexual drive, pattern separation, perception, and cognitive functions, including learning and memory [139, 183–185]. While impaired neurogenesis during fetal development has been linked to intellectual disability disorders, the progressive decline in the neuroblast population followed by the loss of new neurons in the hippocampus has been established as brain aging and distinct neuropathogenic event along with progressive memory loss [47, 139, 144, 184–187]. However, the role of aberrant neurogenesis in pathogenic mechanisms in neuropsychiatric and neurodegenerative disorders has been less explored.
Notably, neurological diseases like epilepsy and cerebral stroke are associated with increased neurogenesis in the hippocampus, unusual migration of neuroblasts in the cortex and striatum, and altered cell fate events in the neurogenic and non-neurogenic areas [48, 51]. A significant percentage of subjects with epileptic seizures and cerebral ischemia have been reported to experience hallucination [188–190]. Besides, physical exercise in a physiological state has been known to enhance cognitive ability by increasing the neurogenesis in the hippocampus [191]. However, aggressive physical exercise appears to exacerbate the disease progression in some neurodegenerative diseases and some individuals experience hallucination after vigorous physical activities [192]. Interestingly, increased level of neuroblast proliferation and their ectopic migration have also become evident in the early phase of neurodegenerative disorders, while the late phase of these disorders with the prominent sign of dementia and depression-related disease have been characterized by diminished levels of neuroblasts in the brain [29, 46, 47, 49, 143, 193]. The ongoing turnover of neuroblasts has generally been confined to the hippocampus and SVZ-OB system, but several reports indicate the possibility for the occurrence and migration of neuroblasts in the other brain regions including the cortex, striatum, and hypothalamus [46, 49, 194–196]. As the astrocytes with radial glial morphology have been identified as the precursors of neuroblasts in the normal brain, the presence of astroglial cells in the non-neurogenic areas has been regarded as an alternate source of NSCs that can give rise to neuroblasts in different areas of the brain during the disease course [197]. Recently, the sustaining of non-newly generated terminally undifferentiated functional neuroblasts has also been reported in the brains of some mammals such as dolphins and sheep [198, 199]. These quiescent neuroblasts are likely to be activated upon environmental stimuli or during disease progression [29, 200]. In disease conditions, the abnormal discharge of proinflammatory cytokine from activated immune cells leads to a cell cycle arrest in NSCs which can cause the stimulation of neuroblast proliferation as a compensatory cellular effect [29, 47]. The concept of reactive neuroblastosis has recently been introduced to describe the increased number of neuroblasts in response to neuropathogenic changes in HD. It describes an abnormal cellular condition of the brain in which neuroblasts undergo extensive proliferation and ectopic migration in the non-canonical neurogenic areas [44, 46]. As the concept is evolving presently, the evidence for the occurrence of reactive neuroblastosis can be correlated with the amount of newly originating neurons/neuroblasts that are positive for markers such as DCX, polysialylated neuronal cell adhesion molecule (PSA-NCAM), neurogenic differentiation (NeuroD), NRG-1, and Calretinin in the brain during the disease conditions [201].
Notably, postmortem studies have identified reduced cell proliferation due to low numbers of kiel (Ki)-67 and proliferating cell nuclear antigen (PCNA) immunopositive cells in the hippocampus of schizophrenia victims [202]. Feng et al. indicated that there is no change in mRNA expression of neuroblast markers in schizophrenic brains compared to age-matched controls indicating a compensatory cellular mechanism upon the aging process accounting for enhanced interstitial white matter neurons [203]. Zhang et al. 2002 unveiled no significant differences in the density of calretinin-positive cells in the mid-dentate gyrus implying that reduced NSC proliferation could be compensated by increased neuroblast in the brain of schizophrenia subjects [204, 205]. However, the reduction in the overall cell proliferation did not reflect the number of neuroblasts in the neurogenic area of the brain, as D Barbeau et al., reported that reduction in the number of PSA-NCAM-immunoreactive neuroblasts was confined in the hilar region but not in the hippocampal dentate gyrus (DG) of brains of schizophrenic subjects [206, 207]. These findings suggest a dynamic balance between NSC proliferation and neuroblast production in the pathophysiology of schizophrenia. Interestingly, a recent report from Joen-Rong Sheu et al. in 2019 revealed an increased number of DCX-expressing neuroblasts with enhanced dendritic arborization, indicating the surplus amount of interacting neuroblasts in the circuit of the hippocampus in the brains of the maternal immune-activated rodent model of schizophrenia [43]. In corroboration with experimental animal study, they demonstrated an increased level of calretinin-positive cells in the hippocampal DG of postmortem brains of schizophrenia [43]. Notably, an immunohistochemical-based study by Walton et al., reported that the numbers of DCX and calretinin-positive cells were increased in the hippocampal DG of the calcium/calmodulin-dependent protein kinase IIα heterozygous knockout (CaMKIIα-hKO) mouse model of Schizophrenia [204, 208]. Further, they have validated these findings by demonstrating the presence of an increased number of calretinin-positive cells in hippocampal DG of post-mortem brain samples from schizophrenia patients compared to control human brains [204]. Eventually, a large number of postmortem data indicated an increased density of interstitial white matter neurons (IWMNs) in the brains with schizophrenia, which could have been derived from the enhanced production of neuroblasts [209].
Chondroitin sulfate proteoglycan (CSPG) is a key constituent of stem cell niches and an important factor for neuronal differentiation, migration of neuroblasts, and synaptogenesis in the adult brain [210]. Strikingly, studies demonstrated increased expression of CSPGs like neural cell adhesion molecules in the brains of subjects with schizophrenia [136]. Besides, increased levels of NRG1, a key component that interacts with ECM have been observed in postmortem brain tissues and induced pluripotent stem cells (iPSCs)-derived neurons of subjects with schizophrenia [211]. NRG-1 is a crucial molecule that facilitates cell fate determination and neuronal differentiation in NSCs via the ErbB4-mediated signaling cascade [212]. Moreover, elevated levels of growth factors, neurotrophic agents, and cytokines, which play key roles in cell fate in neural progenitor cells and facilitate the differentiation of neuroblasts, can serve as indicative measures of reactive neuroblastosis [213]. Notably, pleiotropic growth factors like transforming growth factor (TGF)-beta and metabolic modulators such as peroxisome proliferator-activated receptor (PPAR)-γ have been demonstrated to enhance the number of immature neurons, while their expressions are differential altered in subjects with schizophrenia [29, 47, 213–216]. Hong, S. et al. provide experimental evidence that Poly (ADP-ribose) polymerase (PARP)-1 is required for the differentiation of NSCs, while its absence results in defective neurogenesis and behavioral impairments leading to schizophrenia-like phenotype in experimental animal models [217]. Eventually, altered levels of Wnt signaling components that induce cell cycle events are evident in schizophrenia [75]. A study conducted using iPSCs reported altered Wnt-1 signaling activity in association with abnormal proliferation of neural progenitor cells (NPCs) and altered differentiation of excitatory and inhibitory neurons leading to neuronal circuit miswiring as the developmental origin of schizophrenia [218]. Moreover, subjects with brain metastatic conditions, a state with the malignant proliferation of neuroblasts have been reported to display hallucinations [219]. The animal model of schizophrenia induced by hallucinogenic compounds such as Ketamine and MK-801 (Dizocilpine) has been characterized by an increase in both the quantity and activation of immature neurons in the hippocampus [147, 220]. Notably, the treatment of experimental animals with M108 resulted in an increased amount of neuroblasts and aggravated the symptoms of schizophrenia [43]. As proof of the concept, suppression of the neurogenic process during the adolescent period delayed the onset and progression of schizophrenia-like symptoms in experimental rats [43]. Multiple lines of experimental evidence strongly advocate the intricate relationship between neurogenesis and hallucination in schizophrenia pathology [221].
In the brain, the neuroblasts need to integrate into existing circuits and this process is crucial for proper development and function of the brain. If neuroblasts fail to integrate into existing circuits, they may undergo cell death [222]. Therefore, it can be expected that a surplus generation of neuroblasts would form redundant synapses with existing neurons in a competitive nature. Considering their neurophysiological properties, the variations in the integration of neuroblasts with the existing neural network or retraction of their connections would account for abnormal neuroplasticity in the brain thereby, leading to aberrant neuroplasticity and altered consciousness [44, 223]. Strikingly, neuroblasts have been considered a potential source of the engram, possess electrochemical properties, and exhibit synoptical activities even in an immature state [44, 46, 224]. Engrams are considered to be the biophysiochemical factors generated in the brain that are responsible for cognitive functions, perception, and thinking processes [225]. Recently, abnormal activation of engrams has been proposed as an underlying cause of hallucinations [226] A surplus number of immature neurons resulting from reactive neuroblastosis may tend to integrate into the existing neuronal circuit or may form an extra neural connection in a provocative manner leading to abnormal synoptical, and metabolic activation, thereby drastically misshaping the synapse and electrical events, and creating dysregulation in the release of neurotransmission in the cognitive centers of the diseased brain. Therefore, the abnormal formation of synapses reported in schizophrenia could be associated with differential action potential, neurite outgrowth, and abnormal patterns of synaptic connections during reactive neuroblastosis in the brain [227–229]. The abnormal neurogenic process at the level of integrating an exceeding number of neuroblasts can be proposed to induce abnormal maladaptive behaviors specifically hallucination. Therefore, reactive proliferation and dendritic growth of neuroblasts could potentially be associated with the pathophysiology of hallucination in schizophrenia. While the fate and role of reactive neurblastosis remain unveiled, it is notable that the increased number of neuroblasts tends to be drastically reduced in the end stage of the disease in which depletion of neuroblasts may be associated with dementia. Therefore, the differential modulation of neuroblastosis depends upon the pathogenic or environmental stimuli that could be associated with hallucination and dementia in psychiatric and neurodegenerative disorders (Fig 3). In pathogenic conditions like AD, the neuroblasts tend to express the marker of microglial cells, highlighting their immunological role in the brain [46, 230]. Therefore, further investigation into the mechanisms that drive immunogenic and neurogenic characteristics of neuroblasts in neurodegenerative and schizophrenia contexts could provide valuable insights into the complex interplay between hallucinations and memory loss.
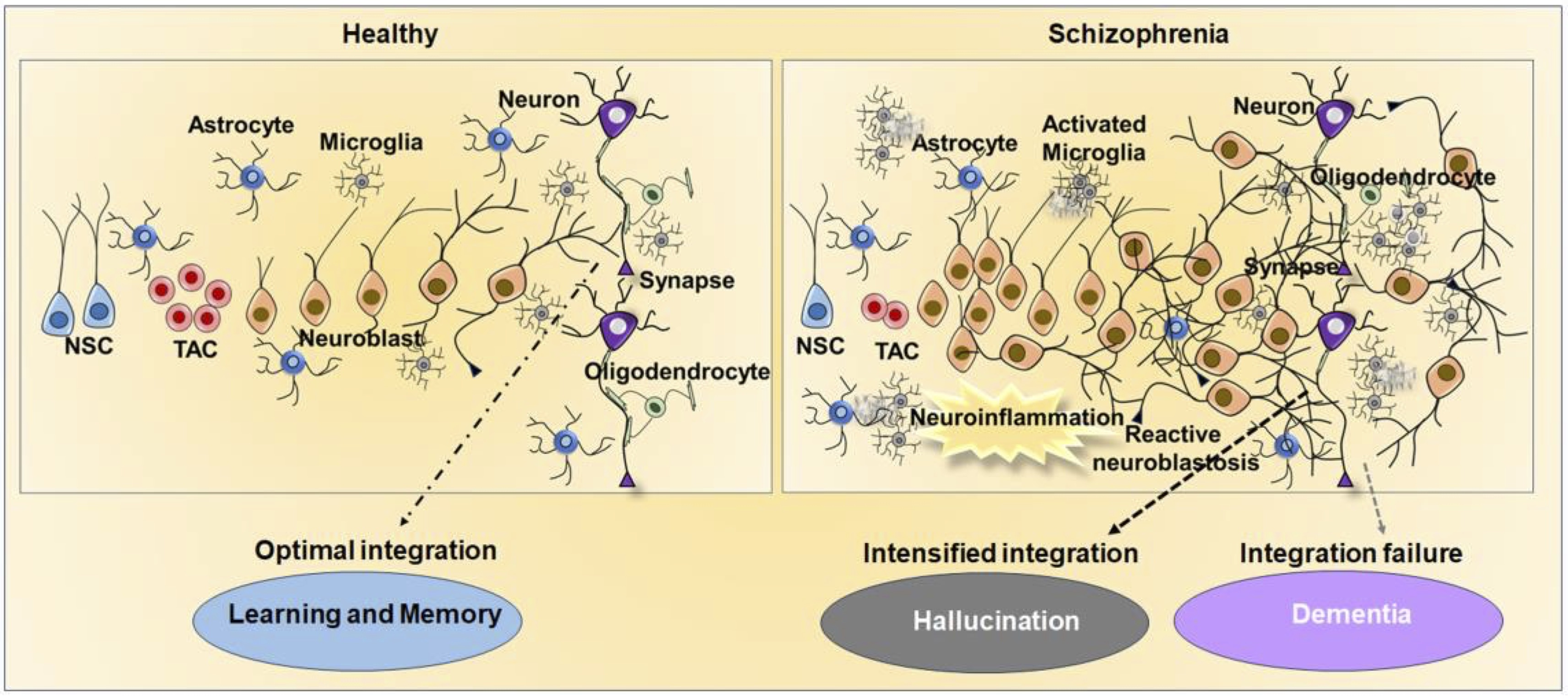
Fig. 3: Regulation of adult neurogenesis and reactive neuroblastosis in health and schizophrenia brains. The picture provides an overview of the neural stem cells (NSC) and transiently amplifying cells (TAC) derived regulation of adult neurogenesis and neuronal integrity in a healthy brain, as well as abnormal neurogenesis and neural integration in schizophrenia. While physiological adult neurogenesis contributes to brain plasticity responsible for cognition function, defective neurogenesis brought on by reactive neuroblastosis causes disruption in neuronal integration and neurotransmission in schizophrenia. Increased neurotransmission from enhanced integration of reactive neuroblasts could be an underlying cause of hallucinations, while decreased neural integration could result in dementia.
Discussion and further direction
The brain is an irreplaceable organ of immense complexity, responsible for synthesizing dreams, imagination, innovative ideas, and a myriad of experiences. Its intricate structure and function majorly rely on neuroregenerative and synaptic plasticity which are driven by both voluntary and involuntary signals. Various functional regions of the brain have a great capacity to spontaneously produce neuroplasticity independent of internal and external stimuli. The key functional areas of the brain have the ability to regenerate throughout life. The regulation of adult neurogenesis mediated by NSCs through the generation of neuroblast have been recognized as a pivotal mechanism underlying diverse cognitive functions, such as intellectual abilities, learning, and memory. Neurogenesis, from development to adulthood, is subject to modulation by a variety of factors. Defects in neurogenesis, particularly at the levels of proliferation and differentiation of NSC and neuroblastosis have been linked to various diseases. Prolonged activation of immune cells can disrupt the neuronal differentiation and integration process of neuroblasts by releasing proinflammatory factors, thereby contributing to cognitive decline, regardless of neuronal loss [34, 47, 151]. Similar to neurodegenerative disorders, schizophrenia has been linked to an aberrant neurogenic process in the brain. Hallucination is the prime behavioral pathology of schizophrenia and subjects with neurological diseases and intake of some drugs induces hallucination for which no confined mechanism has been established. Moreover, the occurrence of dementia has been increasingly evident in schizophrenia [169, 231]. This article emphasizes that reactive neuroblastosis might be responsible for the occurrence of hallucinations as it can abruptly strengthen the synapses and induce abnormal neurotransmission. Besides, failure in the integration of newly generated neuroblasts could contribute to dementia as evidence for neurodegeneration is limited in schizophrenia. Therefore, the futuristic therapeutic targets aimed at harnessing reactive neuroblastosis and correcting the functional integration might be highly beneficial against hallucination and dementia in schizophrenia and other diseases.
With reference to the treatment option, the drugs olanzapine, amisulpride, and ziprasidone have also been implemented to manage the clinical symptoms of schizophrenia and other psychiatric disorders including bipolar disorder. These medications have been linked to the modulation of neurotransmitter systems including serotonin and dopamine which could potentially alter neurogenic process in schizophrenia. Many antipsychotic medications have been shown to have neurotrophic effects by promoting neuronal survival, growth, and synaptic plasticity. For example, preclinical studies have suggested that olanzapine and clozapine enhance BDNF, which could potentially induce neuroblasts to functionally differentiate and effectively integrate into the brain circuit thereby they can harness the reactive neuroblastosis. However, these medications have been known to induce many adverse effects such as movement disorders, seizures, and dementia that are known to be associated with altered regulation of neurogenesis.
Though the turnover of neurogenesis in the human brain and experimental animal brain has been reported to be dissimilar at the level of neuroblast generation, the occurrence of neurogenesis in adulthood has been demonstrated in human brains [184, 232, 233]. In general, understanding the regulation of neurogenesis in the human brain has some drawbacks due to the unavailability of healthy brain samples during the critical period and technical disadvantages [234]. However, the persistence and regulation of neuroblasts have been unequivocally demonstrated in the healthy human brain and aberrant levels of neurogenic process in disease conditions [32, 232, 234, 235]. The reactive neuroblastosis can be investigated using the expression of proliferative markers such as PCNA/Ki67 along with neuroblast markers in postmortem human brains. Eventually, the reporter-based transgenic mouse models that express the marker of neuroblast such as DCX can be subjected to hallucinogenic drugs, and the reactive neuroblastosois can be monitored using live brain imaging and validated by immunohistochemical methods.
Conclusion
Identifying mechanisms underlying hallucination is a fascinating scientific quest in schizophrenia research. While hallucination has been ascertained as the chief psychotic symptom of schizophrenia, the occurrence of hallucination is also increasingly evident in neurodegenerative disorders including AD, PD, and HD. While dementia has generally been attributed to neurodegeneration, recent findings demonstrated that reduced neurogenesis can lead to dementia. Interestingly, schizophrenia has also been characterized by memory loss despite the absence of prominent signs of neurodegeneration. Remarkably, many neurodegenerative disorders have been characterized by reactive neuroblastosis in the progressive pathogenic time frame. While the neuroblasts in the brains have been reported to exhibit electrophysiological properties and serve as a potential cellular resource of engram, abnormal activation of the engrams has been proposed as an underlying cause of hallucination. Therefore, the hypothesis denotes that redundant synaptic connections or their withdrawal from the increased number of neuroblasts may be associated with the generation of abnormal perception resulting in hallucinations. Eventually, failure or defects in the integration of a subset of neuroblasts and their consequent loss could be associated with dementia regardless of the neurodegeneration of existing mature neurons in schizophrenia. While the experimental animal model and few postmortem studies strongly support this hypothesis, adequate human studies are necessary to further validate and understand the role of neuroblastosis in hallucinations. Moreover, the availability of suitable human brain samples, the interference of medication, and methodological drawbacks in assessing the neurogenic process present significant challenges. Presently, the reactive neuroblastosis theory of hallucination provides novel insight into schizophrenia and dementia research, the notion can be further validated with advanced experimental tools and computational modelling. By employing cutting-edge techniques such as high-resolution neuroimaging, optogenetics, and single-cell RNA sequencing, alongside sophisticated bioinformatics tools, a deeper understanding of the mechanisms underlying reactive neuroblastosis can be established. The integrated approach would significantly advance our knowledge of the role of neurogenesis in hallucinations and dementia potentially leading to new therapeutic strategies for psychiatric and neurodegenerative disorders.
Acknowledgements
MK would like to acknowledge and dedicate this article to the late Dr Palani Murugan Rangasamy, Senior Scientist, Centre for Cellular and Molecular Biology (CCMB), Hyderabad, Telangana, India for inspiration. Authors acknowledge UGC-SAP and DST-FIST for the infrastructure support provided to the Department of Animal Science, Bharathidasan University.
Authors contributions
M.K. conceived the concept, hypothesis and contributed to the framework of the manuscript and illustrations. M.P.B.D.I, J.H.M.J, and M.K. wrote the initial draft. All contributed to the revision of the article and made critical comments and suggestions. All authors have read and agreed with this version of the manuscript for submission.
Funding
MK has been supported by the University Grants Commission-Faculty Recharge Programme (UGC-FRP), New Delhi, India. The authors sincerely thank RUSA 2.0, Biological Sciences, Bharathidasan University, (TN RUSA: 311/RUSA (2.0)/2018 dt. 2 December 2020) and Anusandhan National Research Foundation (ANRF)/Science Engineering Research Board (SERB) (CRG/2023/005266), Department of Science and Technology (DST), Government of India for the financial supports.
Ethical approval
Not applicable.
Disclosure Statement
The authors have no conflicts of interest to declare.
References
| 1 | Patel KR, Cherian J, Gohil K, Atkinson D. Schizophrenia: Overview and Treatment Options. P T. 2014 Sep;39(9):638-45.
|
| 2 | Hany M, Rehman B, Azhar Y, Chapman J. Schizophrenia. StatPearls. Treasure Island (FL): StatPearls Publishing; 2023; [cited 2023 Dec 6].Available from: http://www.ncbi.nlm.nih.gov/books/NBK539864/
|
| 3 | Zubin J, Spring B. Vulnerability: A new view of schizophrenia. Journal of Abnormal Psychology. 1977;86(2):103-26.
https://doi.org/10.1037/0021-843X.86.2.103 |
| 4 | Kendler KS. Kraepelin's Final Views on Dementia Praecox. Schizophr Bull. 2020 Dec;47(3):635-43.
https://doi.org/10.1093/schbul/sbaa177 |
| 5 | Ashok AH, Baugh J, Yeragani VK. Paul Eugen Bleuler and the origin of the term schizophrenia (SCHIZOPRENIEGRUPPE). Indian J Psychiatry. 2012;54(1):95-6.
https://doi.org/10.4103/0019-5545.94660 |
| 6 | Correll CU, Schooler NR. Negative Symptoms in Schizophrenia: A Review and Clinical Guide for Recognition, Assessment, and Treatment. Neuropsychiatr Dis Treat. 2020 Feb;16:519-34.
https://doi.org/10.2147/NDT.S225643 |
| 7 | Ruiz-Castañeda P, Santiago Molina E, Aguirre Loaiza H, Daza González MT. Positive symptoms of schizophrenia and their relationship with cognitive and emotional executive functions. Cogn Res Princ Implic. 2022 Aug;7:78.
https://doi.org/10.1186/s41235-022-00428-z |
| 8 | Morrissette DA, Stahl SM. Affective symptoms in schizophrenia. Drug Discovery Today: Therapeutic Strategies. 2011 Jun;8(1):3-9.
https://doi.org/10.1016/j.ddstr.2011.10.005 |
| 9 | Cho W, Shin W-S, An I, Bang M, Cho D-Y, Lee S-H. Biological Aspects of Aggression and Violence in Schizophrenia. Clin Psychopharmacol Neurosci. 2019 Nov;17(4):475-86.
https://doi.org/10.9758/cpn.2019.17.4.475 |
| 10 | McCutcheon RA, Keefe RSE, McGuire PK. Cognitive impairment in schizophrenia: aetiology, pathophysiology, and treatment. Mol Psychiatry. 2023 May;28(5):1902-18.
https://doi.org/10.1038/s41380-023-01949-9 |
| 11 | Schultz SH, North SW, Shields CG. Schizophrenia: a review. Am Fam Physician. 2007 Jun;75(12):1821-9.
|
| 12 | George M, Maheshwari S, Chandran S, Manohar JS, Sathyanarayana Rao TS. Understanding the schizophrenia prodrome. Indian J Psychiatry. 2017;59(4):505-9.
|
| 13 | Saha S, Chant D, Welham J, McGrath J. A Systematic Review of the Prevalence of Schizophrenia. PLoS Med. 2005 May;2(5):e141.
https://doi.org/10.1371/journal.pmed.0020141 |
| 14 | Cederlöf M, Lichtenstein P, Larsson H, Boman M, Rück C, Landén M, et al. Obsessive-Compulsive Disorder, Psychosis, and Bipolarity: A Longitudinal Cohort and Multigenerational Family Study. Schizophr Bull. 2015 Sep;41(5):1076-83.
https://doi.org/10.1093/schbul/sbu169 |
| 15 | Etchecopar-Etchart D, Korchia T, Loundou A, Llorca P-M, Auquier P, Lançon C, et al. Comorbid Major Depressive Disorder in Schizophrenia: A Systematic Review and Meta-Analysis. Schizophr Bull. 2020 Nov;47(2):298-308.
https://doi.org/10.1093/schbul/sbaa153 |
| 16 | Karczewski KJ, Snyder MP. Integrative omics for health and disease. Nat Rev Genet. 2018 May;19(5):299-310.
https://doi.org/10.1038/nrg.2018.4 |
| 17 | Dennison CA, Legge SE, Pardiñas AF, Walters JTR. Genome-wide association studies in schizophrenia: Recent advances, challenges and future perspective. Schizophr Res. 2020 Mar;217:4-12.
https://doi.org/10.1016/j.schres.2019.10.048 |
| 18 | Powell SK, O'Shea CP, Shannon SR, Akbarian S, Brennand KJ. Investigation of Schizophrenia with Human Induced Pluripotent Stem Cells. Adv Neurobiol. 2020;25:155-206.
https://doi.org/10.1007/978-3-030-45493-7_6 |
| 19 | Stępnicki P, Kondej M, Kaczor AA. Current Concepts and Treatments of Schizophrenia. Molecules. 2018 Aug;23(8):2087.
https://doi.org/10.3390/molecules23082087 |
| 20 | Emsley R, Chiliza B, Asmal L, Harvey BH. The nature of relapse in schizophrenia. BMC Psychiatry. 2013 Feb;13:50.
https://doi.org/10.1186/1471-244X-13-50 |
| 21 | Stroup TS, Gray N. Management of common adverse effects of antipsychotic medications. World Psychiatry. 2018 Oct;17(3):341-56.
https://doi.org/10.1002/wps.20567 |
| 22 | Kim R, Healey KL, Sepulveda-Orengo MT, Reissner KJ. Astroglial correlates of neuropsychiatric disease: from Astrocytopathy to Astrogliosis. Prog Neuropsychopharmacol Biol Psychiatry. 2018 Dec;87(Pt A):126-46.
https://doi.org/10.1016/j.pnpbp.2017.10.002 |
| 23 | Abazyan S, Yang EJ, Abazyan B, Xia M, Yang C, Rojas C, et al. Mutant Disrupted-In-Schizophrenia 1 in astrocytes: focus on glutamate metabolism. J Neurosci Res. 2014 Dec;92(12):1659-68.
https://doi.org/10.1002/jnr.23459 |
| 24 | Kolomeets NS [Astroglia of the hippocampus in schizophrenia].. Zh Nevrol Psikhiatr Im S S Korsakova. 2008;108(4):70-6.
|
| 25 | Feresten AH, Barakauskas V, Ypsilanti A, Barr AM, Beasley CL. Increased expression of glial fibrillary acidic protein in prefrontal cortex in psychotic illness. Schizophr Res. 2013 Oct;150(1):252-7.
https://doi.org/10.1016/j.schres.2013.07.024 |
| 26 | Laskaris LE, Di Biase MA, Everall I, Chana G, Christopoulos A, Skafidas E, et al. Microglial activation and progressive brain changes in schizophrenia. Br J Pharmacol. 2016 Feb;173(4):666-80.
https://doi.org/10.1111/bph.13364 |
| 27 | Vallée A. Neuroinflammation in Schizophrenia: The Key Role of the WNT/β-Catenin Pathway. Int J Mol Sci. 2022 Mar;23(5):2810.
https://doi.org/10.3390/ijms23052810 |
| 28 | Radhakrishnan RK, Kandasamy M. SARS-CoV-2-Mediated Neuropathogenesis, Deterioration of Hippocampal Neurogenesis and Dementia. Am J Alzheimers Dis Other Demen. 2022;37:15333175221078418.
https://doi.org/10.1177/15333175221078418 |
| 29 | Kandasamy M, Couillard-Despres S, Raber KA, Stephan M, Lehner B, Winner B, et al. Stem cell quiescence in the hippocampal neurogenic niche is associated with elevated transforming growth factor-beta signaling in an animal model of Huntington disease. J Neuropathol Exp Neurol. 2010 Jul;69(7):717-28.
https://doi.org/10.1097/NEN.0b013e3181e4f733 |
| 30 | Ekdahl CT, Kokaia Z, Lindvall O. Brain inflammation and adult neurogenesis: The dual role of microglia. Neuroscience. 2009 Feb;158(3):1021-9.
https://doi.org/10.1016/j.neuroscience.2008.06.052 |
| 31 | Rao JS, Kellom M, Kim H-W, Rapoport SI. Neuroinflammation and synaptic loss. Neurochem Res. 2012 May;37(5):903-10.
https://doi.org/10.1007/s11064-012-0708-2 |
| 32 | Moreno-Jiménez EP, Flor-García M, Terreros-Roncal J, Rábano A, Cafini F, Pallas-Bazarra N, et al. Adult hippocampal neurogenesis is abundant in neurologically healthy subjects and drops sharply in patients with Alzheimer's disease. Nat Med. 2019 Apr;25(4):554-60.
https://doi.org/10.1038/s41591-019-0375-9 |
| 33 | Marxreiter F, Regensburger M, Winkler J. Adult neurogenesis in Parkinson's disease. Cell Mol Life Sci. 2013 Feb;70(3):459-73.
https://doi.org/10.1007/s00018-012-1062-x |
| 34 | Winner B, Kohl Z, Gage FH. Neurodegenerative disease and adult neurogenesis. Eur J Neurosci. 2011 Mar;33(6):1139-51.
https://doi.org/10.1111/j.1460-9568.2011.07613.x |
| 35 | Eisch AJ, Cameron HA, Encinas JM, Meltzer LA, Ming G-L, Overstreet-Wadiche LS. Adult neurogenesis, mental health, and mental illness: hope or hype? J Neurosci. 2008 Nov;28(46):11785-91.
https://doi.org/10.1523/JNEUROSCI.3798-08.2008 |
| 36 | Apple DM, Fonseca RS, Kokovay E. The role of adult neurogenesis in psychiatric and cognitive disorders. Brain Res. 2017 Jan;1655:270-6.
https://doi.org/10.1016/j.brainres.2016.01.023 |
| 37 | Reif A, Fritzen S, Finger M, Strobel A, Lauer M, Schmitt A, et al. Neural stem cell proliferation is decreased in schizophrenia, but not in depression. Mol Psychiatry. 2006 May;11(5):514-22.
https://doi.org/10.1038/sj.mp.4001791 |
| 38 | Reif A, Schmitt A, Fritzen S, Lesch K-P. Neurogenesis and schizophrenia: dividing neurons in a divided mind? Eur Arch Psychiatry Clin Neurosci. 2007 Aug;257(5):290-9.
https://doi.org/10.1007/s00406-007-0733-3 |
| 39 | Jin K, Peel AL, Mao XO, Xie L, Cottrell BA, Henshall DC, et al. Increased hippocampal neurogenesis in Alzheimer's disease. Proc Natl Acad Sci U S A. 2004 Jan;101(1):343-7.
https://doi.org/10.1073/pnas.2634794100 |
| 40 | Boekhoorn K, Joels M, Lucassen PJ. Increased proliferation reflects glial and vascular-associated changes, but not neurogenesis in the presenile Alzheimer hippocampus. Neurobiol Dis. 2006 Oct;24(1):1-14.
https://doi.org/10.1016/j.nbd.2006.04.017 |
| 41 | Terreros-Roncal J, Moreno-Jiménez EP, Flor-García M, Rodríguez-Moreno CB, Trinchero MF, Cafini F, et al. Impact of neurodegenerative diseases on human adult hippocampal neurogenesis. Science. 2021 Nov;374(6571):1106-13.
https://doi.org/10.1126/science.abl5163 |
| 42 | Ruzo A, Croft GF, Metzger JJ, Galgoczi S, Gerber LJ, Pellegrini C, et al. Chromosomal instability during neurogenesis in Huntington's disease. Development. 2018 Jan;145(2):dev156844.
https://doi.org/10.1242/dev.156844 |
| 43 | Sheu J-R, Hsieh C-Y, Jayakumar T, Tseng M-F, Lee H-N, Huang S-W, et al. A Critical Period for the Development of Schizophrenia-Like Pathology by Aberrant Postnatal Neurogenesis. Front Neurosci. 2019;13:635.
https://doi.org/10.3389/fnins.2019.00635 |
| 44 | Kandasamy M, Aigner L. Reactive Neuroblastosis in Huntington's Disease: A Putative Therapeutic Target for Striatal Regeneration in the Adult Brain. Front Cell Neurosci. 2018;12:37.
https://doi.org/10.3389/fncel.2018.00037 |
| 45 | Couillard-Despres S, Winner B, Schaubeck S, Aigner R, Vroemen M, Weidner N, et al. Doublecortin expression levels in adult brain reflect neurogenesis. Eur J Neurosci. 2005 Jan;21(1):1-14.
https://doi.org/10.1111/j.1460-9568.2004.03813.x |
| 46 | Kandasamy M, Aigner L. Neuroplasticity, limbic neuroblastosis and neuro-regenerative disorders. Neural Regen Res. 2018 Aug;13(8):1322-6.
https://doi.org/10.4103/1673-5374.235214 |
| 47 | Kandasamy M, Anusuyadevi M, Aigner KM, Unger MS, Kniewallner KM, de Sousa DMB, et al. TGF-β Signaling: A Therapeutic Target to Reinstate Regenerative Plasticity in Vascular Dementia? Aging Dis. 2020 Jul;11(4):828-50.
https://doi.org/10.14336/AD.2020.0222 |
| 48 | Lindvall O, Kokaia Z. Neurogenesis following Stroke Affecting the Adult Brain. Cold Spring Harb Perspect Biol. 2015 Nov;7(11):a019034.
https://doi.org/10.1101/cshperspect.a019034 |
| 49 | Kandasamy M, Rosskopf M, Wagner K, Klein B, Couillard-Despres S, Reitsamer HA, et al. Reduction in subventricular zone-derived olfactory bulb neurogenesis in a rat model of Huntington's disease is accompanied by striatal invasion of neuroblasts. PLoS One. 2015;10(2):e0116069.
https://doi.org/10.1371/journal.pone.0116069 |
| 50 | Zheng W, ZhuGe Q, Zhong M, Chen G, Shao B, Wang H, et al. Neurogenesis in Adult Human Brain after Traumatic Brain Injury. J Neurotrauma. 2013 Nov;30(22):1872-80.
https://doi.org/10.1089/neu.2010.1579 |
| 51 | Jessberger S, Parent JM. Epilepsy and Adult Neurogenesis. Cold Spring Harb Perspect Biol. 2015 Dec;7(12):a020677.
https://doi.org/10.1101/cshperspect.a020677 |
| 52 | Roshan SA, Elangovan G, Gunaseelan D, Jayachandran SK, Kandasamy M, Anusuyadevi M. Pathogenomic Signature and Aberrant Neurogenic Events in Experimental Cerebral Ischemic Stroke: A Neurotranscriptomic-Based Implication for Dementia. J Alzheimers Dis. 2023;94(s1):S289-308.
https://doi.org/10.3233/JAD-220831 |
| 53 | EL HAJ M, ROCHE J, JARDRI R, KAPOGIANNIS D, GALLOUJ K, ANTOINE P. Clinical and neurocognitive aspects of hallucinations in Alzheimer's disease. Neurosci Biobehav Rev. 2017 Dec;83:713-20.
https://doi.org/10.1016/j.neubiorev.2017.02.021 |
| 54 | Poewe W. When a Parkinson's disease patient starts to hallucinate. Pract Neurol. 2008 Aug;8(4):238-41.
https://doi.org/10.1136/jnnp.2008.152579 |
| 55 | Rosenblatt A. Neuropsychiatry of Huntington's disease. Dialogues Clin Neurosci. 2007 Jun;9(2):191-7.
https://doi.org/10.31887/DCNS.2007.9.2/arosenblatt |
| 56 | Walker E, Kestler L, Bollini A, Hochman KM. Schizophrenia: etiology and course. Annu Rev Psychol. 2004;55:401-30.
https://doi.org/10.1146/annurev.psych.55.090902.141950 |
| 57 | Gejman PV, Sanders AR, Duan J. The role of genetics in the etiology of schizophrenia. Psychiatr Clin North Am. 2010 Mar;33(1):35-66.
https://doi.org/10.1016/j.psc.2009.12.003 |
| 58 | McCutcheon RA, Reis Marques T, Howes OD. Schizophrenia-An Overview. JAMA Psychiatry. 2020 Feb;77(2):201-10.
https://doi.org/10.1001/jamapsychiatry.2019.3360 |
| 59 | Tandon R, Keshavan MS, Nasrallah HA. Schizophrenia, "just the facts" what we know in 2008. 2. Epidemiology and etiology. Schizophr Res. 2008 Jul;102(1-3):1-18.
https://doi.org/10.1016/j.schres.2008.04.011 |
| 60 | Stilo SA, Murray RM. Non-Genetic Factors in Schizophrenia. Curr Psychiatry Rep. 2019 Sep;21(10):100.
https://doi.org/10.1007/s11920-019-1091-3 |
| 61 | Fatemi SH, Folsom TD. The Neurodevelopmental Hypothesis of Schizophrenia, Revisited. Schizophr Bull. 2009 May;35(3):528-48.
https://doi.org/10.1093/schbul/sbn187 |
| 62 | O'Connell P, Woodruff PW, Wright I, Jones P, Murray RM. Developmental insanity or dementia praecox: was the wrong concept adopted? Schizophr Res. 1997 Feb;23(2):97-106.
https://doi.org/10.1016/S0920-9964(96)00110-7 |
| 63 | Hartenstein V, Stollewerk A. The Evolution of Early Neurogenesis. Dev Cell. 2015 Feb;32(4):390-407.
https://doi.org/10.1016/j.devcel.2015.02.004 |
| 64 | Brown AS, Derkits EJ. Prenatal Infection and Schizophrenia: A Review of Epidemiologic and Translational Studies. Am J Psychiatry. 2010 Mar;167(3):261-80.
https://doi.org/10.1176/appi.ajp.2009.09030361 |
| 65 | Muraki K, Tanigaki K. Neuronal migration abnormalities and its possible implications for schizophrenia. Front Neurosci. 2015;9:74.
https://doi.org/10.3389/fnins.2015.00074 |
| 66 | Gupta S, Kulhara P. What is schizophrenia: A neurodevelopmental or neurodegenerative disorder or a combination of both? A critical analysis. Indian J Psychiatry. 2010;52(1):21-7.
https://doi.org/10.4103/0019-5545.58891 |
| 67 | Nadarajah B, Alifragis P, Wong ROL, Parnavelas JG. Neuronal migration in the developing cerebral cortex: observations based on real-time imaging. Cereb Cortex. 2003 Jun;13(6):607-11.
https://doi.org/10.1093/cercor/13.6.607 |
| 68 | Buchsbaum IY, Cappello S. Neuronal migration in the CNS during development and disease: insights from in vivo and in vitro models. Development. 2019 Jan;146(1):dev163766.
https://doi.org/10.1242/dev.163766 |
| 69 | Tee JY, Mackay-Sim A. Directional Persistence of Cell Migration in Schizophrenia Patient-Derived Olfactory Cells. Int J Mol Sci. 2021 Aug;22(17):9177.
https://doi.org/10.3390/ijms22179177 |
| 70 | Goo BS, Mun DJ, Kim S, Nhung TTM, Lee SB, Woo Y, et al. Schizophrenia-associated Mitotic Arrest Deficient-1 (MAD1) regulates the polarity of migrating neurons in the developing neocortex. Mol Psychiatry. 2023 Feb;28(2):856-70.
https://doi.org/10.1038/s41380-022-01856-5 |
| 71 | Năstase MG, Vlaicu I, Trifu SC. Genetic polymorphism and neuroanatomical changes in schizophrenia. Rom J Morphol Embryol. 2022;63(2):307-22.
https://doi.org/10.47162/RJME.63.2.03 |
| 72 | Tomita K, Kubo K, Ishii K, Nakajima K. Disrupted-in-Schizophrenia-1 (Disc1) is necessary for migration of the pyramidal neurons during mouse hippocampal development. Hum Mol Genet. 2011 Jul;20(14):2834-45.
https://doi.org/10.1093/hmg/ddr194 |
| 73 | Duan X, Chang JH, Ge S, Faulkner RL, Kim JY, Kitabatake Y, et al. Disrupted-In-Schizophrenia 1 regulates integration of newly generated neurons in the adult brain. Cell. 2007 Sep;130(6):1146-58.
https://doi.org/10.1016/j.cell.2007.07.010 |
| 74 | Hayashi K, Kubo K, Kitazawa A, Nakajima K. Cellular dynamics of neuronal migration in the hippocampus. Frontiers in Neuroscience. 2015 [cited 2024 Feb 1]. ;9. Available from: https://www.frontiersin.org/articles/10.3389/fnins.2015.00135
https://doi.org/10.3389/fnins.2015.00135 |
| 75 | Hoseth EZ, Krull F, Dieset I, Mørch RH, Hope S, Gardsjord ES, et al. Exploring the Wnt signaling pathway in schizophrenia and bipolar disorder. Transl Psychiatry. 2018 Mar;8(1):1-10.
https://doi.org/10.1038/s41398-018-0102-1 |
| 76 | McGuire JL, Depasquale EA, Funk AJ, O'Donnovan SM, Hasselfeld K, Marwaha S, et al. Abnormalities of signal transduction networks in chronic schizophrenia. NPJ Schizophr. 2017 Sep;3:30.
https://doi.org/10.1038/s41537-017-0032-6 |
| 77 | Okazaki S, Boku S, Otsuka I, Mouri K, Aoyama S, Shiroiwa K, et al. The cell cycle-related genes as biomarkers for schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry. 2016 Oct;70:85-91.
https://doi.org/10.1016/j.pnpbp.2016.05.005 |
| 78 | Javitt DC. Glutamate and schizophrenia: phencyclidine, N-methyl-D-aspartate receptors, and dopamine-glutamate interactions. Int Rev Neurobiol. 2007;78:69-108.
https://doi.org/10.1016/S0074-7742(06)78003-5 |
| 79 | Beck K, Hindley G, Borgan F, Ginestet C, McCutcheon R, Brugger S, et al. Association of Ketamine With Psychiatric Symptoms and Implications for Its Therapeutic Use and for Understanding Schizophrenia: A Systematic Review and Meta-analysis. JAMA Netw Open. 2020 May;3(5):e204693.
https://doi.org/10.1001/jamanetworkopen.2020.4693 |
| 80 | Dietz AG, Goldman SA, Nedergaard M. Glial cells in schizophrenia: A unified hypothesis. Lancet Psychiatry. 2020 Mar;7(3):272-81.
https://doi.org/10.1016/S2215-0366(19)30302-5 |
| 81 | Brisch R, Saniotis A, Wolf R, Bielau H, Bernstein H-G, Steiner J, et al. The Role of Dopamine in Schizophrenia from a Neurobiological and Evolutionary Perspective: Old Fashioned, but Still in Vogue. Front Psychiatry. 2014 May;5:47.
https://doi.org/10.3389/fpsyt.2014.00110 |
| 82 | Davis KL, Kahn RS, Ko G, Davidson M. Dopamine in schizophrenia: a review and reconceptualization. Am J Psychiatry. 1991 Nov;148(11):1474-86.
https://doi.org/10.1176/ajp.148.11.1474 |
| 83 | Tripathi PP, Bozzi Y. The role of dopaminergic and serotonergic systems in neurodevelopmental disorders: a focus on epilepsy and seizure susceptibility. Bioimpacts. 2015;5(2):97-102.
https://doi.org/10.15171/bi.2015.07 |
| 84 | Kokkinou M, Ashok AH, Howes OD. The effects of ketamine on dopaminergic function: meta-analysis and review of the implications for neuropsychiatric disorders. Mol Psychiatry. 2018 Jan;23(1):59-69.
https://doi.org/10.1038/mp.2017.190 |
| 85 | Jentsch JD, Roth RH. The neuropsychopharmacology of phencyclidine: from NMDA receptor hypofunction to the dopamine hypothesis of schizophrenia. Neuropsychopharmacology. 1999 Mar;20(3):201-25.
https://doi.org/10.1016/S0893-133X(98)00060-8 |
| 86 | Xu M, Wong AHC. GABAergic inhibitory neurons as therapeutic targets for cognitive impairment in schizophrenia. Acta Pharmacol Sin. 2018 May;39(5):733-53.
https://doi.org/10.1038/aps.2017.172 |
| 87 | Scarr E, Gibbons AS, Neo J, Udawela M, Dean B. Cholinergic connectivity: it's implications for psychiatric disorders. Front Cell Neurosci. 2013 May;7:55.
https://doi.org/10.3389/fncel.2013.00055 |
| 88 | Mäki-Marttunen V, Andreassen OA, Espeseth T. The role of norepinephrine in the pathophysiology of schizophrenia. Neurosci Biobehav Rev. 2020 Nov;118:298-314.
https://doi.org/10.1016/j.neubiorev.2020.07.038 |
| 89 | Salleh MR. The Genetics of Schizophrenia. Malays J Med Sci. 2004 Jul;11(2):3-11.
|
| 90 | McDonald C, Murphy KC. The new genetics of schizophrenia. Psychiatr Clin North Am. 2003 Mar;26(1):41-63.
https://doi.org/10.1016/S0193-953X(02)00030-8 |
| 91 | de la Serna E, Baeza I, Andrés S, Puig O, Sánchez-Guistau V, Romero S, et al. Comparison between young siblings and offspring of subjects with schizophrenia: clinical and neuropsychological characteristics. Schizophr Res. 2011 Sep;131(1-3):35-42.
https://doi.org/10.1016/j.schres.2011.06.015 |
| 92 | Stefansson H, Sigurdsson E, Steinthorsdottir V, Bjornsdottir S, Sigmundsson T, Ghosh S, et al. Neuregulin 1 and Susceptibility to Schizophrenia. Am J Hum Genet. 2002 Oct;71(4):877-92.
https://doi.org/10.1086/342734 |
| 93 | Williams NM, Preece A, Morris DW, Spurlock G, Bray NJ, Stephens M, et al. Identification in 2 independent samples of a novel schizophrenia risk haplotype of the dystrobrevin binding protein gene (DTNBP1). Arch Gen Psychiatry. 2004 Apr;61(4):336-44.
https://doi.org/10.1001/archpsyc.61.4.336 |
| 94 | Ho B-C, Wassink TH, O'Leary DS, Sheffield VC, Andreasen NC. Catechol-O-methyl transferase Val158Met gene polymorphism in schizophrenia: working memory, frontal lobe MRI morphology and frontal cerebral blood flow. Mol Psychiatry. 2005 Mar;10(3):229, 287-98.
https://doi.org/10.1038/sj.mp.4001616 |
| 95 | Wise CD, Stein L. Dopamine-beta-hydroxylase deficits in the brains of schizophrenic patients. Science. 1973 Jul;181(4097):344-7.
https://doi.org/10.1126/science.181.4097.344 |
| 96 | Mirnics K, Middleton FA, Stanwood GD, Lewis DA, Levitt P. Disease-specific changes in regulator of G-protein signaling 4 (RGS4) expression in schizophrenia. Mol Psychiatry. 2001 May;6(3):293-301.
https://doi.org/10.1038/sj.mp.4000866 |
| 97 | Hodgkinson CA, Goldman D, Jaeger J, Persaud S, Kane JM, Lipsky RH, et al. Disrupted in Schizophrenia 1 (DISC1): Association with Schizophrenia, Schizoaffective Disorder, and Bipolar Disorder. The American Journal of Human Genetics. 2004 Nov;75(5):862-72.
https://doi.org/10.1086/425586 |
| 98 | De Luca V, Tharmalingam S, Zai C, Potapova N, Strauss J, Vincent J, et al. Association of HPA axis genes with suicidal behaviour in schizophrenia. J Psychopharmacol. 2010 May;24(5):677-82.
https://doi.org/10.1177/0269881108097817 |
| 99 | Matthysse S, Sugarman J. Neurotransmitter Theories of Schizophrenia. In: Iversen LL, Iversen SD, Snyder SH, editors. Handbook of Psychopharmacology: Volume 10: Neuroleptics and Schizophrenia. Boston, MA: Springer US; 1978; pp 221-42.
https://doi.org/10.1007/978-1-4613-4042-3_7 |
| 100 | Jacobi W, Winkler H. Encephalographische Studien an chronisch Schizophrenen. Archiv f Psychiatrie. 1927 Dec;81(1):299-332.
https://doi.org/10.1007/BF01825649 |
| 101 | Ellis JK, Walker EF, Goldsmith DR. Selective Review of Neuroimaging Findings in Youth at Clinical High Risk for Psychosis: On the Path to Biomarkers for Conversion. Front Psychiatry. 2020 Sep;11:567534.
https://doi.org/10.3389/fpsyt.2020.567534 |
| 102 | Johnstone EC, Crow TJ, Frith CD, Husband J, Kreel L. Cerebral ventricular size and cognitive impairment in chronic schizophrenia. Lancet. 1976 Oct;2(7992):924-6.
https://doi.org/10.1016/S0140-6736(76)90890-4 |
| 103 | Vita A, De Peri L, Deste G, Sacchetti E. Progressive loss of cortical gray matter in schizophrenia: a meta-analysis and meta-regression of longitudinal MRI studies. Transl Psychiatry. 2012 Nov;2(11):e190.
https://doi.org/10.1038/tp.2012.116 |
| 104 | Cropley VL, Klauser P, Lenroot RK, Bruggemann J, Sundram S, Bousman C, et al. Accelerated Gray and White Matter Deterioration With Age in Schizophrenia. Am J Psychiatry. 2017 Mar;174(3):286-95.
https://doi.org/10.1176/appi.ajp.2016.16050610 |
| 105 | Valdés-Tovar M, Rodríguez-Ramírez AM, Rodríguez-Cárdenas L, Sotelo-Ramírez CE, Camarena B, Sanabrais-Jiménez MA, et al. Insights into myelin dysfunction in schizophrenia and bipolar disorder. World J Psychiatry. 2022 Feb;12(2):264-85.
https://doi.org/10.5498/wjp.v12.i2.264 |
| 106 | Apostolova LG, Green AE, Babakchanian S, Hwang KS, Chou Y-Y, Toga AW, et al. Hippocampal atrophy and ventricular enlargement in normal aging, mild cognitive impairment and Alzheimer's disease. Alzheimer Dis Assoc Disord. 2012 Jan;26(1):17-27.
https://doi.org/10.1097/WAD.0b013e3182163b62 |
| 107 | Mak E, Su L, Williams GB, Firbank MJ, Lawson RA, Yarnall AJ, et al. Longitudinal whole-brain atrophy and ventricular enlargement in nondemented Parkinson's disease. Neurobiol Aging. 2017 Jul;55:78-90.
https://doi.org/10.1016/j.neurobiolaging.2017.03.012 |
| 108 | Horga G, Bernacer J, Dusi N, Entis J, Chu K, Hazlett EA, et al. Correlations between ventricular enlargement and gray and white matter volumes of cortex, thalamus, striatum, and internal capsule in schizophrenia. Eur Arch Psychiatry Clin Neurosci. 2011;261(7):467-76.
https://doi.org/10.1007/s00406-011-0202-x |
| 109 | Gaser C, Nenadic I, Buchsbaum BR, Hazlett EA, Buchsbaum MS. Ventricular enlargement in schizophrenia related to volume reduction of the thalamus, striatum, and superior temporal cortex. Am J Psychiatry. 2004 Jan;161(1):154-6.
https://doi.org/10.1176/appi.ajp.161.1.154 |
| 110 | Selemon LD, Goldman-Rakic PS. The reduced neuropil hypothesis: a circuit based model of schizophrenia. Biol Psychiatry. 1999 Jan;45(1):17-25.
https://doi.org/10.1016/S0006-3223(98)00281-9 |
| 111 | Osimo EF, Beck K, Reis Marques T, Howes OD. Synaptic loss in schizophrenia: a meta-analysis and systematic review of synaptic protein and mRNA measures. Mol Psychiatry. 2019 Apr;24(4):549-61.
https://doi.org/10.1038/s41380-018-0041-5 |
| 112 | Selemon LD, Rajkowska G, Goldman-Rakic PS. Elevated neuronal density in prefrontal area 46 in brains from schizophrenic patients: application of a three-dimensional, stereologic counting method. J Comp Neurol. 1998 Mar;392(3):402-12.
https://doi.org/10.1002/(SICI)1096-9861(19980316)392:3<402::AID-CNE9>3.3.CO;2-3 |
| 113 | Najjar S, Pearlman DM. Neuroinflammation and white matter pathology in schizophrenia: systematic review. Schizophrenia Research. 2015 Jan;161(1):102-12.
https://doi.org/10.1016/j.schres.2014.04.041 |
| 114 | Sprooten E, Papmeyer M, Smyth AM, Vincenz D, Honold S, Conlon GA, et al. Cortical thickness in first-episode schizophrenia patients and individuals at high familial risk: a cross-sectional comparison. Schizophr Res. 2013 Dec;151(1-3):259-64.
https://doi.org/10.1016/j.schres.2013.09.024 |
| 115 | Sheffield JM, Barch DM. Cognition and resting-state functional connectivity in schizophrenia. Neuroscience & Biobehavioral Reviews. 2016 Feb;61:108-20.
https://doi.org/10.1016/j.neubiorev.2015.12.007 |
| 116 | Jiang S, Huang H, Zhou J, Li H, Duan M, Yao D, et al. Progressive trajectories of schizophrenia across symptoms, genes, and the brain. BMC Med. 2023 Jul;21:237.
https://doi.org/10.1186/s12916-023-02935-2 |
| 117 | Liu Y, Ouyang P, Zheng Y, Mi L, Zhao J, Ning Y, et al. A Selective Review of the Excitatory-Inhibitory Imbalance in Schizophrenia: Underlying Biology, Genetics, Microcircuits, and Symptoms. Front Cell Dev Biol. 2021 Oct;9:664535.
https://doi.org/10.3389/fcell.2021.664535 |
| 118 | Turetsky B, Cowell PE, Gur RC, Grossman RI, Shtasel DL, Gur RE. Frontal and temporal lobe brain volumes in schizophrenia. Relationship to symptoms and clinical subtype. Arch Gen Psychiatry. 1995 Dec;52(12):1061-70.
https://doi.org/10.1001/archpsyc.1995.03950240079013 |
| 119 | Kaur A, Basavanagowda DM, Rathod B, Mishra N, Fuad S, Nosher S, et al. Structural and Functional Alterations of the Temporal lobe in Schizophrenia: A Literature Review. Cureus. 12(10):e11177.
|
| 120 | Jaaro-Peled H, Ayhan Y, Pletnikov MV, Sawa A. Review of Pathological Hallmarks of Schizophrenia: Comparison of Genetic Models With Patients and Nongenetic Models. Schizophr Bull. 2010 Mar;36(2):301-13.
https://doi.org/10.1093/schbul/sbp133 |
| 121 | Glausier JR, Lewis DA. Dendritic Spine Pathology in Schizophrenia. Neuroscience. 2013 Oct;251:90-107.
https://doi.org/10.1016/j.neuroscience.2012.04.044 |
| 122 | Arnold SJM, Ivleva EI, Gopal TA, Reddy AP, Jeon-Slaughter H, Sacco CB, et al. Hippocampal Volume Is Reduced in Schizophrenia and Schizoaffective Disorder But Not in Psychotic Bipolar I Disorder Demonstrated by Both Manual Tracing and Automated Parcellation (FreeSurfer). Schizophr Bull. 2015 Jan;41(1):233-49.
https://doi.org/10.1093/schbul/sbu009 |
| 123 | Heckers S, Konradi C. Hippocampal neurons in schizophrenia. J Neural Transm. 2002 May;109(0):891-905.
https://doi.org/10.1007/s007020200073 |
| 124 | Sweet RA, Pierri JN, Auh S, Sampson AR, Lewis DA. Reduced pyramidal cell somal volume in auditory association cortex of subjects with schizophrenia. Neuropsychopharmacology. 2003 Mar;28(3):599-609.
https://doi.org/10.1038/sj.npp.1300120 |
| 125 | Sasabayashi D, Yoshimura R, Takahashi T, Takayanagi Y, Nishiyama S, Higuchi Y, et al. Reduced Hippocampal Subfield Volume in Schizophrenia and Clinical High-Risk State for Psychosis. Front Psychiatry. 2021 Mar;12:642048.
https://doi.org/10.3389/fpsyt.2021.642048 |
| 126 | Gemmell E, Bosomworth H, Allan L, Hall R, Khundakar A, Oakley AE, et al. Hippocampal Neuronal Atrophy and Cognitive Function in Delayed Poststroke and Aging-Related Dementias. Stroke. 2012 Mar;43(3):808-14.
https://doi.org/10.1161/STROKEAHA.111.636498 |
| 127 | Uysal G, Ozturk M. Hippocampal atrophy based Alzheimer's disease diagnosis via machine learning methods. Journal of Neuroscience Methods. 2020 May;337:108669.
https://doi.org/10.1016/j.jneumeth.2020.108669 |
| 128 | Pirildar S, Gönül AS, Taneli F, Akdeniz F. Low serum levels of brain-derived neurotrophic factor in patients with schizophrenia do not elevate after antipsychotic treatment. Prog Neuropsychopharmacol Biol Psychiatry. 2004 Jul;28(4):709-13.
https://doi.org/10.1016/j.pnpbp.2004.05.008 |
| 129 | Ho B-C, Andreasen NC, Dawson JD, Wassink TH. Association between brain-derived neurotrophic factor Val66Met gene polymorphism and progressive brain volume changes in schizophrenia. Am J Psychiatry. 2007 Dec;164(12):1890-9.
https://doi.org/10.1176/appi.ajp.2007.05111903 |
| 130 | Rizos EN, Papathanasiou M, Michalopoulou PG, Mazioti A, Douzenis A, Kastania A, et al. Association of serum BDNF levels with hippocampal volumes in first psychotic episode drug-naive schizophrenic patients. Schizophrenia Research. 2011 Jul;129(2):201-4.
https://doi.org/10.1016/j.schres.2011.03.011 |
| 131 | Erickson KI, Prakash RS, Voss MW, Chaddock L, Heo S, McLaren M, et al. Brain-Derived Neurotrophic Factor Is Associated with Age-Related Decline in Hippocampal Volume. J Neurosci. 2010 Apr;30(15):5368-75.
https://doi.org/10.1523/JNEUROSCI.6251-09.2010 |
| 132 | Iritani S, Niizato K, Nawa H, Ikeda K, Emson PC. Immunohistochemical study of brain-derived neurotrophic factor and its receptor, TrkB, in the hippocampal formation of schizophrenic brains. Prog Neuropsychopharmacol Biol Psychiatry. 2003 Aug;27(5):801-7.
https://doi.org/10.1016/S0278-5846(03)00112-X |
| 133 | Takahashi M, Shirakawa O, Toyooka K, Kitamura N, Hashimoto T, Maeda K, et al. Abnormal expression of brain-derived neurotrophic factor and its receptor in the corticolimbic system of schizophrenic patients. Mol Psychiatry. 2000 May;5(3):293-300.
https://doi.org/10.1038/sj.mp.4000718 |
| 134 | Weickert CS, Hyde TM, Lipska BK, Herman MM, Weinberger DR, Kleinman JE. Reduced brain-derived neurotrophic factor in prefrontal cortex of patients with schizophrenia. Mol Psychiatry. 2003 Jun;8(6):592-610.
https://doi.org/10.1038/sj.mp.4001308 |
| 135 | Soles A, Selimovic A, Sbrocco K, Ghannoum F, Hamel K, Moncada EL, et al. Extracellular Matrix Regulation in Physiology and in Brain Disease. Int J Mol Sci. 2023 Apr;24(8):7049.
https://doi.org/10.3390/ijms24087049 |
| 136 | Berretta S. Extracellular Matrix Abnormalities in Schizophrenia. Neuropharmacology. 2012 Mar;62(3):1584-97.
https://doi.org/10.1016/j.neuropharm.2011.08.010 |
| 137 | Bitanihirwe BKY, Woo T-UW. Perineuronal Nets and Schizophrenia: The Importance of Neuronal Coatings. Neurosci Biobehav Rev. 2014 Sep;45:85-99.
https://doi.org/10.1016/j.neubiorev.2014.03.018 |
| 138 | Shah A, Lodge DJ. A loss of hippocampal perineuronal nets produces deficits in dopamine system function: relevance to the positive symptoms of schizophrenia. Transl Psychiatry. 2013 Jan;3(1):e215-e215.
https://doi.org/10.1038/tp.2012.145 |
| 139 | Toda T, Parylak SL, Linker SB, Gage FH. The role of adult hippocampal neurogenesis in brain health and disease. Mol Psychiatry. 2019 Jan;24(1):67-87.
https://doi.org/10.1038/s41380-018-0036-2 |
| 140 | Kempermann G, Song H, Gage FH. Neurogenesis in the Adult Hippocampus. Cold Spring Harb Perspect Biol. 2015 Sep;7(9):a018812.
https://doi.org/10.1101/cshperspect.a018812 |
| 141 | Lieberman JA, Girgis RR, Brucato G, Moore H, Provenzano F, Kegeles L, et al. Hippocampal dysfunction in the pathophysiology of schizophrenia: a selective review and hypothesis for early detection and intervention. Mol Psychiatry. 2018 Aug;23(8):1764-72.
https://doi.org/10.1038/mp.2017.249 |
| 142 | Pujol N, Penadés R, Junqué C, Dinov I, Fu CHY, Catalán R, et al. Hippocampal abnormalities and age in chronic schizophrenia: morphometric study across the adult lifespan. Br J Psychiatry. 2014 Nov;205(5):369-75.
https://doi.org/10.1192/bjp.bp.113.140384 |
| 143 | Poornimai Abirami GP, Radhakrishnan RK, Johnson E, Roshan SA, Yesudhas A, Parveen S, et al. The Regulation of Reactive Neuroblastosis, Neuroplasticity, and Nutraceuticals for Effective Management of Autism Spectrum Disorder. Adv Neurobiol. 2020;24:207-22.
https://doi.org/10.1007/978-3-030-30402-7_8 |
| 144 | Babcock KR, Page JS, Fallon JR, Webb AE. Adult Hippocampal Neurogenesis in Aging and Alzheimer's Disease. Stem Cell Reports. 2021 Apr;16(4):681-93.
https://doi.org/10.1016/j.stemcr.2021.01.019 |
| 145 | Hagihara H, Takao K, Walton NM, Matsumoto M, Miyakawa T. Immature Dentate Gyrus: An Endophenotype of Neuropsychiatric Disorders. Neural Plast. 2013;2013:318596.
https://doi.org/10.1155/2013/318596 |
| 146 | Tavitian A, Song W, Schipper HM. Dentate Gyrus Immaturity in Schizophrenia. Neuroscientist. 2019 Dec;25(6):528-47.
https://doi.org/10.1177/1073858418824072 |
| 147 | Rawat R, Tunc-Ozcan E, McGuire TL, Peng C-Y, Kessler JA. Ketamine activates adult-born immature granule neurons to rapidly alleviate depression-like behaviors in mice. Nat Commun. 2022 May;13(1):2650.
https://doi.org/10.1038/s41467-022-30386-5 |
| 148 | Mao Y, Ge X, Frank CL, Madison JM, Koehler AN, Doud MK, et al. Disrupted in schizophrenia 1 regulates neuronal progenitor proliferation via modulation of GSK3beta/beta-catenin signaling. Cell. 2009 Mar;136(6):1017-31.
https://doi.org/10.1016/j.cell.2008.12.044 |
| 149 | Liu J, Suzuki T, Seki T, Namba T, Tanimura A, Arai H. Effects of repeated phencyclidine administration on adult hippocampal neurogenesis in the rat. Synapse. 2006 Jul;60(1):56-68.
https://doi.org/10.1002/syn.20275 |
| 150 | Burghaus L, Eggers C, Timmermann L, Fink GR, Diederich NJ. Hallucinations in Neurodegenerative Diseases. CNS Neurosci Ther. 2011 Feb;18(2):149-59.
https://doi.org/10.1111/j.1755-5949.2011.00247.x |
| 151 | Winner B, Winkler J. Adult Neurogenesis in Neurodegenerative Diseases. Cold Spring Harb Perspect Biol. 2015 Apr;7(4):a021287.
https://doi.org/10.1101/cshperspect.a021287 |
| 152 | Boksa P. On the neurobiology of hallucinations. J Psychiatry Neurosci. 2009 Jul;34(4):260-2.
|
| 153 | Hare SM. Hallucinations: A Functional Network Model of How Sensory Representations Become Selected for Conscious Awareness in Schizophrenia. Front Neurosci. 2021 Nov;15:733038.
https://doi.org/10.3389/fnins.2021.733038 |
| 154 | Jardri R, Pouchet A, Pins D, Thomas P. Cortical activations during auditory verbal hallucinations in schizophrenia: a coordinate-based meta-analysis. Am J Psychiatry. 2011 Jan;168(1):73-81.
https://doi.org/10.1176/appi.ajp.2010.09101522 |
| 155 | Wu JL, Haberman RP, Gallagher M, Koh MT. Probing for Conditioned Hallucinations Through Neural Activation in a Ketamine Mouse Model of Schizophrenia. Neurosci Bull. 2020 May;36(8):937-41.
https://doi.org/10.1007/s12264-020-00507-5 |
| 156 | Telles-Correia D, Moreira AL, Gonçalves JS. Hallucinations and related concepts-their conceptual background. Front Psychol. 2015 Jul;6:991.
https://doi.org/10.3389/fpsyg.2015.00991 |
| 157 | Andreasen NC. Concept of Schizophrenia: Past, Present, and Future. Schizophrenia. John Wiley & Sons, Ltd; 2010; pp 1-8.
https://doi.org/10.1002/9781444327298.ch1 |
| 158 | Ibor JJL. Lecciones de psicologia medica: según apuntes tomados en la cátedra. Lecciones de psicologia medica: según apuntes tomados en la cátedra. 1964; [cited 2023 Dec 10]; pp 427-427.
|
| 159 | Feinberg I. Corollary Discharge, Hallucinations, and Dreaming. Schizophr Bull. 2011 Jan;37(1):1-3.
https://doi.org/10.1093/schbul/sbq115 |
| 160 | Jackson H. The Selected Writings of John Hughlings Jackson. Volume 1. On Epilepsy and Epileptiform Convulsions. Archives of Neurology & Psychiatry. 1932 Mar;27(3):757.
https://doi.org/10.1001/archneurpsyc.1932.02230150273022 |
| 161 | Penfield W, Boldrey E. SOMATIC MOTOR AND SENSORY REPRESENTATION IN THE CEREBRAL CORTEX OF MAN AS STUDIED BY ELECTRICAL STIMULATION. Brain. 1937;60(4):389-443.
https://doi.org/10.1093/brain/60.4.389 |
| 162 | Catani M. A little man of some importance. Brain. 2017 Nov;140(11):3055-61.
https://doi.org/10.1093/brain/awx270 |
| 163 | Perez-Costas E, Melendez-Ferro M, Roberts RC. BASAL GANGLIA PATHOLOGY IN SCHIZOPHRENIA: DOPAMINE CONNECTIONS and ANOMALIES. J Neurochem. 2010 Apr;113(2):287-302.
https://doi.org/10.1111/j.1471-4159.2010.06604.x |
| 164 | Kumar S, Soren S, Chaudhury S. Hallucinations: Etiology and clinical implications. Ind Psychiatry J. 2009;18(2):119-26.
https://doi.org/10.4103/0972-6748.62273 |
| 165 | Khandaker GM, Cousins L, Deakin J, Lennox BR, Yolken R, Jones PB. Inflammation and immunity in schizophrenia: implications for pathophysiology and treatment. Lancet Psychiatry. 2015 Mar;2(3):258-70.
https://doi.org/10.1016/S2215-0366(14)00122-9 |
| 166 | McGuire PK, Shah GM, Murray RM. Increased blood flow in Broca's area during auditory hallucinations in schizophrenia. Lancet. 1993 Sep;342(8873):703-6.
https://doi.org/10.1016/0140-6736(93)91707-S |
| 167 | Wang Q, Ren H, Li C, Li Z, Li J, Li H, et al. Metabolite differences in the medial prefrontal cortex in schizophrenia patients with and without persistent auditory verbal hallucinations: a 1H MRS study. Transl Psychiatry. 2022 Mar;12(1):1-9.
https://doi.org/10.1038/s41398-022-01866-5 |
| 168 | Cleghorn JM, Garnett ES, Nahmias C, Brown GM, Kaplan RD, Szechtman H, et al. Regional brain metabolism during auditory hallucinations in chronic schizophrenia. Br J Psychiatry. 1990 Oct;157:562-70.
https://doi.org/10.1192/bjp.157.4.562 |
| 169 | Chaudhury S. Hallucinations: Clinical aspects and management. Ind Psychiatry J. 2010;19(1):5-12.
https://doi.org/10.4103/0972-6748.77625 |
| 170 | Degueure A, Fontenot A, Husan A, Khan MW. An Unusual Presentation of Vivid Hallucinations. Cureus. 14(5):e25441.
|
| 171 | Gottlieb JD, Mueser KT, Rosenberg SD, Xie H, Wolfe RS. Psychotic Depression, Posttraumatic Stress Disorder, and Engagement in Cognitive-Behavioral Therapy within an Outpatient Sample of Adults with Serious Mental Illness. Compr Psychiatry. 2011;52(1):41-9.
https://doi.org/10.1016/j.comppsych.2010.04.012 |
| 172 | Bassiony MM, Lyketsos CG. Delusions and hallucinations in Alzheimer's disease: review of the brain decade. Psychosomatics. 2003;44(5):388-401.
https://doi.org/10.1176/appi.psy.44.5.388 |
| 173 | Patel SS, Attard A, Jacobsen P, Shergill S. Acetylcholinesterase Inhibitors (AChEI's) for the treatment of visual hallucinations in schizophrenia: a review of the literature. BMC Psychiatry. 2010 Sep;10:69.
https://doi.org/10.1186/1471-244X-10-69 |
| 174 | Barnes J, David AS. Visual hallucinations in Parkinson's disease: a review and phenomenological survey. J Neurol Neurosurg Psychiatry. 2001 Jun;70(6):727-33.
https://doi.org/10.1136/jnnp.70.6.727 |
| 175 | Lafay-Chebassier C, Chavant F, Favrelière S, Pizzoglio V, Pérault-Pochat M-C, French Association of Regional Pharmacovigilance Centers. Drug-induced Depression: a Case/Non Case Study in the French Pharmacovigilance Database. Therapie. 2015;70(5):425-32.
https://doi.org/10.2515/therapie/2015026 |
| 176 | Niebrzydowska A, Grabowski J. Medication-induced Psychotic Disorder. A Review of Selected Drugs Side Effects. Psychiatr Danub. 2022;34(1):11-8.
https://doi.org/10.24869/psyd.2022.11 |
| 177 | Rolland B, Jardri R, Amad A, Thomas P, Cottencin O, Bordet R. Pharmacology of hallucinations: several mechanisms for one single symptom? Biomed Res Int. 2014;2014:307106.
https://doi.org/10.1155/2014/307106 |
| 178 | Taoufik E, Kouroupi G, Zygogianni O, Matsas R. Synaptic dysfunction in neurodegenerative and neurodevelopmental diseases: an overview of induced pluripotent stem-cell-based disease models. Open Biol. 2018 Sep;8(9):180138.
https://doi.org/10.1098/rsob.180138 |
| 179 | Prasad T, Iyer S, Chatterjee S, Kumar M. In vivo models to study neurogenesis and associated neurodevelopmental disorders-Microcephaly and autism spectrum disorder. WIREs Mechanisms of Disease. 2023;15(4):e1603.
https://doi.org/10.1002/wsbm.1603 |
| 180 | Ahmad R, Sportelli V, Ziller M, Spengler D, Hoffmann A. Tracing Early Neurodevelopment in Schizophrenia with Induced Pluripotent Stem Cells. Cells. 2018 Sep;7(9):140.
https://doi.org/10.3390/cells7090140 |
| 181 | Toro CT, Deakin JFW. Adult neurogenesis and schizophrenia: A window on abnormal early brain development? Schizophrenia Research. 2007 Feb;90(1):1-14.
https://doi.org/10.1016/j.schres.2006.09.030 |
| 182 | Iannitelli A, Quartini A, Tirassa P, Bersani G. Schizophrenia and neurogenesis: A stem cell approach. Neurosci Biobehav Rev. 2017 Sep;80:414-42.
https://doi.org/10.1016/j.neubiorev.2017.06.010 |
| 183 | Snyder JS, Drew MR. Functional Neurogenesis Over the Years. Behav Brain Res. 2020 Mar;382:112470.
https://doi.org/10.1016/j.bbr.2020.112470 |
| 184 | Ming G, Song H. Adult Neurogenesis in the Mammalian Brain: Significant Answers and Significant Questions. Neuron. 2011 May;70(4):687-702.
https://doi.org/10.1016/j.neuron.2011.05.001 |
| 185 | Kempermann G, Wiskott L, Gage FH. Functional significance of adult neurogenesis. Curr Opin Neurobiol. 2004 Apr;14(2):186-91.
https://doi.org/10.1016/j.conb.2004.03.001 |
| 186 | Stagni F, Giacomini A, Emili M, Guidi S, Bartesaghi R. Neurogenesis impairment: An early developmental defect in Down syndrome. Free Radic Biol Med. 2018 Jan;114:15-32.
https://doi.org/10.1016/j.freeradbiomed.2017.07.026 |
| 187 | Guarnieri FC, de Chevigny A, Falace A, Cardoso C. Disorders of neurogenesis and cortical development. Dialogues Clin Neurosci. 2018 Dec;20(4):255-66.
https://doi.org/10.31887/DCNS.2018.20.4/ccardoso |
| 188 | Stangeland H, Orgeta V, Bell V. Poststroke psychosis: a systematic review. J Neurol Neurosurg Psychiatry. 2018 Aug;89(8):879-85.
https://doi.org/10.1136/jnnp-2017-317327 |
| 189 | Cascella NG, Schretlen DJ, Sawa A. SCHIZOPHRENIA AND EPILEPSY: IS THERE A SHARED SUSCEPTIBILITY? Neurosci Res. 2009 Apr;63(4):227-35.
https://doi.org/10.1016/j.neures.2009.01.002 |
| 190 | Kasper BS, Kasper EM, Pauli E, Stefan H. Phenomenology of hallucinations, illusions, and delusions as part of seizure semiology. Epilepsy Behav. 2010 May;18(1-2):13-23.
https://doi.org/10.1016/j.yebeh.2010.03.006 |
| 191 | Liu PZ, Nusslock R. Exercise-Mediated Neurogenesis in the Hippocampus via BDNF. Front Neurosci. 2018 Feb;12:52.
https://doi.org/10.3389/fnins.2018.00052 |
| 192 | Lessell S. Exercise-Induced Visual Hallucinations A Symptom of Occipital Lobe Tumors. Journal of Neuro-Ophthalmology. 1988 Jun;8(2):81.
|
| 193 | Manickam N, Radhakrishnan RK, Vergil Andrews JF, Selvaraj DB, Kandasamy M. Cell cycle re-entry of neurons and reactive neuroblastosis in Huntington's disease: Possibilities for neural-glial transition in the brain. Life Sci. 2020 Dec;263:118569.
https://doi.org/10.1016/j.lfs.2020.118569 |
| 194 | Bartkowska K, Turlejski K, Koguc-Sobolewska P, Djavadian R. Adult Neurogenesis in the Mammalian Hypothalamus: Impact of Newly Generated Neurons on Hypothalamic Function. Neuroscience. 2023 Apr;515:83-92.
https://doi.org/10.1016/j.neuroscience.2023.02.012 |
| 195 | Kandasamy M, Radhakrishnan RK, Poornimai Abirami GP, Roshan SA, Yesudhas A, Balamuthu K, et al. Possible Existence of the Hypothalamic-Pituitary-Hippocampal (HPH) Axis: A Reciprocal Relationship Between Hippocampal Specific Neuroestradiol Synthesis and Neuroblastosis in Ageing Brains with Special Reference to Menopause and Neurocognitive Disorders. Neurochem Res. 2019 Aug;44(8):1781-95.
https://doi.org/10.1007/s11064-019-02833-1 |
| 196 | Jurkowski MP, Bettio L, K. Woo E, Patten A, Yau S-Y, Gil-Mohapel J. Beyond the Hippocampus and the SVZ: Adult Neurogenesis Throughout the Brain. Front Cell Neurosci. 2020 Sep;14:576444.
https://doi.org/10.3389/fncel.2020.576444 |
| 197 | Gonzalez-Perez O, Quiñones-Hinojosa A. Astrocytes as neural stem cells in the adult brain. J Stem Cells. 2012;7(3):181-8.
https://doi.org/10.1155/2012/378356 |
| 198 | Parolisi R, Cozzi B, Bonfanti L. Humans and Dolphins: Decline and Fall of Adult Neurogenesis. Front Neurosci. 2018 Jul;12:497.
https://doi.org/10.3389/fnins.2018.00497 |
| 199 | Piumatti M, Palazzo O, La Rosa C, Crociara P, Parolisi R, Luzzati F, et al. Non-Newly Generated, "Immature" Neurons in the Sheep Brain Are Not Restricted to Cerebral Cortex. J Neurosci. 2018 Jan;38(4):826-42.
https://doi.org/10.1523/JNEUROSCI.1781-17.2017 |
| 200 | Urbán N, Cheung TH. Stem cell quiescence: the challenging path to activation. Development. 2021 Feb;148(3):dev165084.
https://doi.org/10.1242/dev.165084 |
| 201 | von Bohlen und Halbach O. Immunohistological markers for proliferative events, gliogenesis, and neurogenesis within the adult hippocampus. Cell Tissue Res. 2011 Jul;345(1):1-19.
https://doi.org/10.1007/s00441-011-1196-4 |
| 202 | Allen KM, Fung SJ, Shannon Weickert C. Cell proliferation is reduced in the hippocampus in schizophrenia. Aust N Z J Psychiatry. 2016 May;50(5):473-80.
https://doi.org/10.1177/0004867415589793 |
| 203 | Fung SJ, Joshi D, Allen KM, Sivagnanasundaram S, Rothmond DA, Saunders R, et al. Developmental Patterns of Doublecortin Expression and White Matter Neuron Density in the Postnatal Primate Prefrontal Cortex and Schizophrenia. PLoS One. 2011 Sep;6(9):e25194.
https://doi.org/10.1371/journal.pone.0025194 |
| 204 | Walton NM, Zhou Y, Kogan JH, Shin R, Webster M, Gross AK, et al. Detection of an immature dentate gyrus feature in human schizophrenia/bipolar patients. Transl Psychiatry. 2012 Jul;2(7):e135.
https://doi.org/10.1038/tp.2012.56 |
| 205 | Zhang ZJ, Reynolds GP. A selective decrease in the relative density of parvalbumin-immunoreactive neurons in the hippocampus in schizophrenia. Schizophr Res. 2002 May;55(1-2):1-10.
https://doi.org/10.1016/S0920-9964(01)00188-8 |
| 206 | Weissleder C, North HF, Shannon Weickert C. Important unanswered questions about adult neurogenesis in schizophrenia. Curr Opin Psychiatry. 2019 May;32(3):170-8.
https://doi.org/10.1097/YCO.0000000000000501 |
| 207 | Barbeau D, Liang JJ, Robitalille Y, Quirion R, Srivastava LK. Decreased expression of the embryonic form of the neural cell adhesion molecule in schizophrenic brains. Proc Natl Acad Sci U S A. 1995 Mar;92(7):2785-9.
https://doi.org/10.1073/pnas.92.7.2785 |
| 208 | Walton NM, Shin R, Tajinda K, Heusner CL, Kogan JH, Miyake S, et al. Adult neurogenesis transiently generates oxidative stress. PLoS One. 2012;7(4):e35264.
https://doi.org/10.1371/journal.pone.0035264 |
| 209 | Duchatel RJ, Weickert CS, Tooney PA. White matter neuron biology and neuropathology in schizophrenia. NPJ Schizophrenia. 2019;5. DOI: 10.1038/s41537-019-0078-8
https://doi.org/10.1038/s41537-019-0078-8 |
| 210 | Mencio CP, Hussein RK, Yu P, Geller HM. The Role of Chondroitin Sulfate Proteoglycans in Nervous System Development. J Histochem Cytochem. 2021 Jan;69(1):61-80.
https://doi.org/10.1369/0022155420959147 |
| 211 | Law AJ, Wang Y, Sei Y, O'Donnell P, Piantadosi P, Papaleo F, et al. Neuregulin 1-ErbB4-PI3K signaling in schizophrenia and phosphoinositide 3-kinase-p110δ inhibition as a potential therapeutic strategy. Proceedings of the National Academy of Sciences. 2012 Jul;109(30):12165-70.
https://doi.org/10.1073/pnas.1206118109 |
| 212 | Mei L, Xiong W-C. Neuregulin 1 in neural development, synaptic plasticity and schizophrenia. Nat Rev Neurosci. 2008 Jun;9(6):437-52.
https://doi.org/10.1038/nrn2392 |
| 213 | Kandasamy M, Lehner B, Kraus S, Sander PR, Marschallinger J, Rivera FJ, et al. TGF-beta signalling in the adult neurogenic niche promotes stem cell quiescence as well as generation of new neurons. J Cell Mol Med. 2014 Jul;18(7):1444-59.
https://doi.org/10.1111/jcmm.12298 |
| 214 | Pan S, Zhou Y, Yan L, Xuan F, Tong J, Li Y, et al. TGF-β1 is associated with deficits in cognition and cerebral cortical thickness in first-episode schizophrenia. J Psychiatry Neurosci. 2022 Mar;47(2):E86-98.
https://doi.org/10.1503/jpn.210121 |
| 215 | Cortez I, Denner L, Dineley KT. Divergent Mechanisms for PPARγ Agonism in Ameliorating Aging-Related Versus Cranial Irradiation-Induced Context Discrimination Deficits. Frontiers in Aging Neuroscience. 2019 [cited 2024 May 24]. ;11. Available from: https://www.frontiersin.org/articles/10.3389/fnagi.2019.00038
https://doi.org/10.3389/fnagi.2019.00038 |
| 216 | Sagheddu C, Melis M, Muntoni AL, Pistis M. Repurposing Peroxisome Proliferator-Activated Receptor Agonists in Neurological and Psychiatric Disorders. Pharmaceuticals (Basel). 2021 Oct;14(10):1025.
https://doi.org/10.3390/ph14101025 |
| 217 | Hong S, Yi JH, Lee S, Park C-H, Ryu JH, Shin KS, et al. Defective neurogenesis and schizophrenia-like behavior in PARP-1-deficient mice. Cell Death Dis. 2019 Dec;10(12):1-16.
https://doi.org/10.1038/s41419-019-2174-0 |
| 218 | Topol A, Zhu S, Tran N, Simone A, Fang G, Brennand KJ. Altered WNT signaling in hiPSC NPCs derived from four schizophrenia patients. Biol Psychiatry. 2015 Sep;78(6):e29-34.
https://doi.org/10.1016/j.biopsych.2014.12.028 |
| 219 | Romero-Luna G, Mejía-Pérez SI, Ramírez-Cruz J, Aguilar-Hidalgo KM, Ocampo-Díaz KM, Moscardini-Martelli J, et al. Schizophrenia-Like Psychosis Presented in a Patient With a Temporal Lobe Tumor: A Case Report. Cureus. 14(9):e29034.
|
| 220 | Singh S, Mishra A, Srivastava N, Shukla S. MK-801 (Dizocilpine) Regulates Multiple Steps of Adult Hippocampal Neurogenesis and Alters Psychological Symptoms via Wnt/β-Catenin Signaling in Parkinsonian Rats. ACS Chem Neurosci. 2017 Mar;8(3):592-605.
https://doi.org/10.1021/acschemneuro.6b00354 |
| 221 | Lodge D, Mercier MS. Ketamine and phencyclidine: the good, the bad and the unexpected. Br J Pharmacol. 2015 Sep;172(17):4254-76.
https://doi.org/10.1111/bph.13222 |
| 222 | Shors TJ, Anderson ML, Curlik DM, Nokia MS. Use it or lose it: how neurogenesis keeps the brain fit for learning. Behav Brain Res. 2012 Feb;227(2):450-8.
https://doi.org/10.1016/j.bbr.2011.04.023 |
| 223 | Klempin F, Kronenberg G, Cheung G, Kettenmann H, Kempermann G. Properties of Doublecortin-(DCX)-Expressing Cells in the Piriform Cortex Compared to the Neurogenic Dentate Gyrus of Adult Mice. PLoS ONE. 2011;6(10). DOI: 10.1371/journal.pone.0025760
https://doi.org/10.1371/journal.pone.0025760 |
| 224 | Baptista P, Andrade JP. Adult Hippocampal Neurogenesis: Regulation and Possible Functional and Clinical Correlates. Frontiers in Neuroanatomy. 2018 [cited 2023 Dec 11]. ;12. Available from: https://www.frontiersin.org/articles/10.3389/fnana.2018.00044
https://doi.org/10.3389/fnana.2018.00044 |
| 225 | Ortega-de San Luis C, Ryan TJ. Understanding the physical basis of memory: Molecular mechanisms of the engram. J Biol Chem. 2022 Mar;298(5):101866.
https://doi.org/10.1016/j.jbc.2022.101866 |
| 226 | Bouso JC, Ona G, Kohek M, dos Santos RG, Hallak JEC, Alcázar-Córcoles MÁ, et al. Hallucinations and Hallucinogens: Psychopathology or Wisdom? Cult Med Psychiatry. 2023;47(2):576-604.
https://doi.org/10.1007/s11013-022-09814-0 |
| 227 | Yin D-M, Chen Y-J, Sathyamurthy A, Xiong W-C, Mei L. Synaptic dysfunction in schizophrenia. Adv Exp Med Biol. 2012;970:493-516.
https://doi.org/10.1007/978-3-7091-0932-8_22 |
| 228 | Faludi G, Mirnics K. Synaptic changes in the brain of subjects with schizophrenia. Int J Dev Neurosci. 2011 May;29(3):305-9.
https://doi.org/10.1016/j.ijdevneu.2011.02.013 |
| 229 | OBI-NAGATA K, TEMMA Y, HAYASHI-TAKAGI A. Synaptic functions and their disruption in schizophrenia: From clinical evidence to synaptic optogenetics in an animal model. Proc Jpn Acad Ser B Phys Biol Sci. 2019 May;95(5):179-97.
https://doi.org/10.2183/pjab.95.014 |
| 230 | Unger MS, Marschallinger J, Kaindl J, Klein B, Johnson M, Khundakar AA, et al. Doublecortin expression in CD8+ T-cells and microglia at sites of amyloid-β plaques: A potential role in shaping plaque pathology? Alzheimers Dement. 2018 Aug;14(8):1022-37.
https://doi.org/10.1016/j.jalz.2018.02.017 |
| 231 | Cai L, Huang J. Schizophrenia and risk of dementia: a meta-analysis study. Neuropsychiatr Dis Treat. 2018 Aug;14:2047-55.
https://doi.org/10.2147/NDT.S172933 |
| 232 | Kumar A, Pareek V, Faiq MA, Ghosh SK, Kumari C. ADULT NEUROGENESIS IN HUMANS: A Review of Basic Concepts, History, Current Research, and Clinical Implications. Innov Clin Neurosci. 2019 May;16(5-6):30-7.
|
| 233 | Amrein I. Adult Hippocampal Neurogenesis in Natural Populations of Mammals. Cold Spring Harb Perspect Biol. 2015 May;7(5):a021295.
https://doi.org/10.1101/cshperspect.a021295 |
| 234 | Kempermann G, Gage FH, Aigner L, Song H, Curtis MA, Thuret S, et al. Human Adult Neurogenesis: Evidence and Remaining Questions. Cell Stem Cell. 2018 Jul;23(1):25-30.
https://doi.org/10.1016/j.stem.2018.04.004 |
| 235 | Leal-Galicia P, Chávez-Hernández ME, Mata F, Mata-Luévanos J, Rodríguez-Serrano LM, Tapia-de-Jesús A, et al. Adult Neurogenesis: A Story Ranging from Controversial New Neurogenic Areas and Human Adult Neurogenesis to Molecular Regulation. Int J Mol Sci. 2021 Oct;22(21):11489.
https://doi.org/10.3390/ijms222111489 |