Tracing the Evolution and Recombination Events of Neurotropic Arenaviridae Viruses: a Bioinformatics Approach
bDepartment of Computer Science, College of Science, University of Diyala, Iraq
Keywords
Abstract
Lassa virus is a member of the Arenaviridae family, a major cause of viral hemorrhagic fever. This virus is associated with severe neurological complications in a select group of patients. The evolutionary mechanisms behind genetic diversity, its adaptation, and its potential neuropathogenicity are still poorly understood. In this study, a comprehensive evolutionary analysis of the S and L genomes of Lassavirus was conducted, with an emphasis on ancestral reconstruction and genomic structure, as well as the identification of mutations that may contribute to viral adaptation leading to neurological disease. A set of complete S and L genomes was collected from NCBI. These sequences were carefully aligned using the MUSCLE algorithm to ensure a high match at each site. GTR+Gamma was used for evolutionary inference and was selected based on a statistical model test for its ability to accurately reflect the evolutionary dynamics of Lassa virus genomes. Phylogenetic trees were constructed using the maximum-likelihood algorithm RAxML, followed by careful preliminary analyses to assess the reliability of each branch in the tree. The mutations and recombination at the ancestral node were identified, which is likely a crucial point in the virus's ability to adapt and evolve. The emergence and distribution of major mutations across the viral lineage can be monitored. Notably, strains linked to known neurological problems frequently exhibit mutations, suggesting a possible link between certain genetic alterations and LASV's neuroinvasive characteristics. Our outcomes shed light on how genetic variety in the S and L segments impacts neurotropic virulence and offer important new insights into the evolutionary history and genomic adaptability of LASV. In order to anticipate neurological risk, create centered diagnostics, and direct the establishment of medical methods against neurotropic arenavirus infection, this study sets up the basis for future research.
Introduction
With exponentially increasing genomic and biological data, bioinformatics has emerged as an enabling science of this era, particularly in medicine, genetics, microbiology, and drug discovery. Bioinformatics refers to the application of computer technology for analysing biological data like DNA, RNA, or protein sequences. It enables researchers to manipulate, compare, and analyse vast genomic databases; identify mutations, gene functions, and evolutionary relationships; build phylogenetic trees; reconstruct ancestral sequences; and accelerate drug target discovery and disease gene mapping. Bioinformatics places the scale and complexity of biological science today into manageable parameters [1].
Based on the structure of their nucleic acid genomes, viruses are generally divided into DNA or RNA viruses. DNA viruses had more host specificity and phylogenetic similarity in their genomic sequences compared to RNA viruses. The Arenaviridae family of RNA viruses has varied host species and genomic structures [2].
The nomenclature "Arenavirus" originates from the Latin terms "arenosus," meaning "sandy," and "arena," signifying "sand," due to the "sandy" shape of Arenavirus particles seen under an electron microscope. An arenavirus genome comprises two, and occasionally three, single-stranded RNA segments designated as short (S), medium (M), and large (L) [3].
Zoonotic transmission of certain pathogenic marine viruses to human beings on contact with infected animal cadavers, feces, or material infested with them can cause risky and sometimes fatal diseases with hemorrhagic or neurological features, but natural infection in their hosts is typically asymptomatic [4].
RNA-virus-like pathogens that can cause life-threatening and severe diseases in human beings include viruses of the Arenaviridae family. The most famous of these viruses is viral hemorrhagic fever. This family is geographically divided into Old World viruses (such as Lassa) and New World viruses (such as Junin and Machupo). Lassa virus is considered a major challenge to global public health, as it has caused hundreds of thousands of infections and thousands of deaths [5]. Despite this deadly threat, there is no widely licensed vaccine yet. While earlier studies have mapped out how different Lassa virus strains are related, they only looked at one part of the virus's genetic material (either S or L), missing the possible effects of mixing different parts. This oversight could bias the conclusions, leading to incorrect conclusions about the virus's evolutionary relationships or its history [6].
In addition, the identity of the specific genetic mutations that occurred in the past and enabled the virus to adapt to prevailing conditions and spread remains largely unknown [7], [8]. Ancestral reconstruction techniques are useful because they allow us to look back in time at the molecular level [9]. With the help of these powerful computational tools, the most likely ancestral genome sequences of the virus can be inferred from its historical divergence using statistical and evolutionary methods [10].
Recent studies show that Lassa virus infection expands beyond hemorrhagic and systemic symptoms, which could result in major brain damage in certain patients, involving encephalitis, visual problems, and lasting disorders of the brain [11]. Knowing the familial roots of these neurological illnesses is an essential step to developing suitable diagnostic and medical methods. Analyzing the genomic evolution of the virus, including mutation examination, single-nucleotide polymorphisms (SNPs), and genomic recombination sequences, is necessary for understanding how the virus has adapted to gain new characteristics, including the ability to enter the nervous system.
This research presents the gathering and study of complete genomes of the S and L segments of the Lassa virus from global databases, using modern computational and analytical techniques, involving multiple alignment, optimal gene substitution model selection, phylogeny tree construction, and genomic information ancestry reconstruction. The current research aims to clarify genetic changes and the history of the evolution of the virus while studying the potential link between these changes and the onset of neurological challenges in people with the virus. The conclusions of this research project add immensely to our knowledge of neuroviral development and may contribute to the detection, management, and avoidance of viral diseases that impact the body's nervous system.
This research aims to improve our understanding by creating evolutionary trees for the genomes of the two groups (L and S) using the maximum likelihood algorithm, and the following sections will describe how the oldest common ancestor of the virus genomes was built.
Materials and Methods
Complete S and L segment genomes of Lassa virus were obtained from NCBI, with duplicates and incomplete entries removed [12]. Multiple sequence alignment was performed using MUSCLE, and the best-fit substitution model (GTR+Gamma) was selected. Phylogenetic trees were reconstructed with the maximum-likelihood method in RAxML, supported by bootstrap analysis. Single nucleotide polymorphisms (SNPs) and mutations were identified by comparative alignment, and ancestral genomes were reconstructed using RAxML-NG to infer evolutionary trends. Full technical details (software parameters, command-line instructions, preprocessing scripts) are available in the Supplementary Methods. This section delineates a series of fundamental research steps, each deemed crucial and essential for attaining optimal outcomes. The following sections will explain the comprehensive steps involved in the research process, as illustrated in Fig. 1.
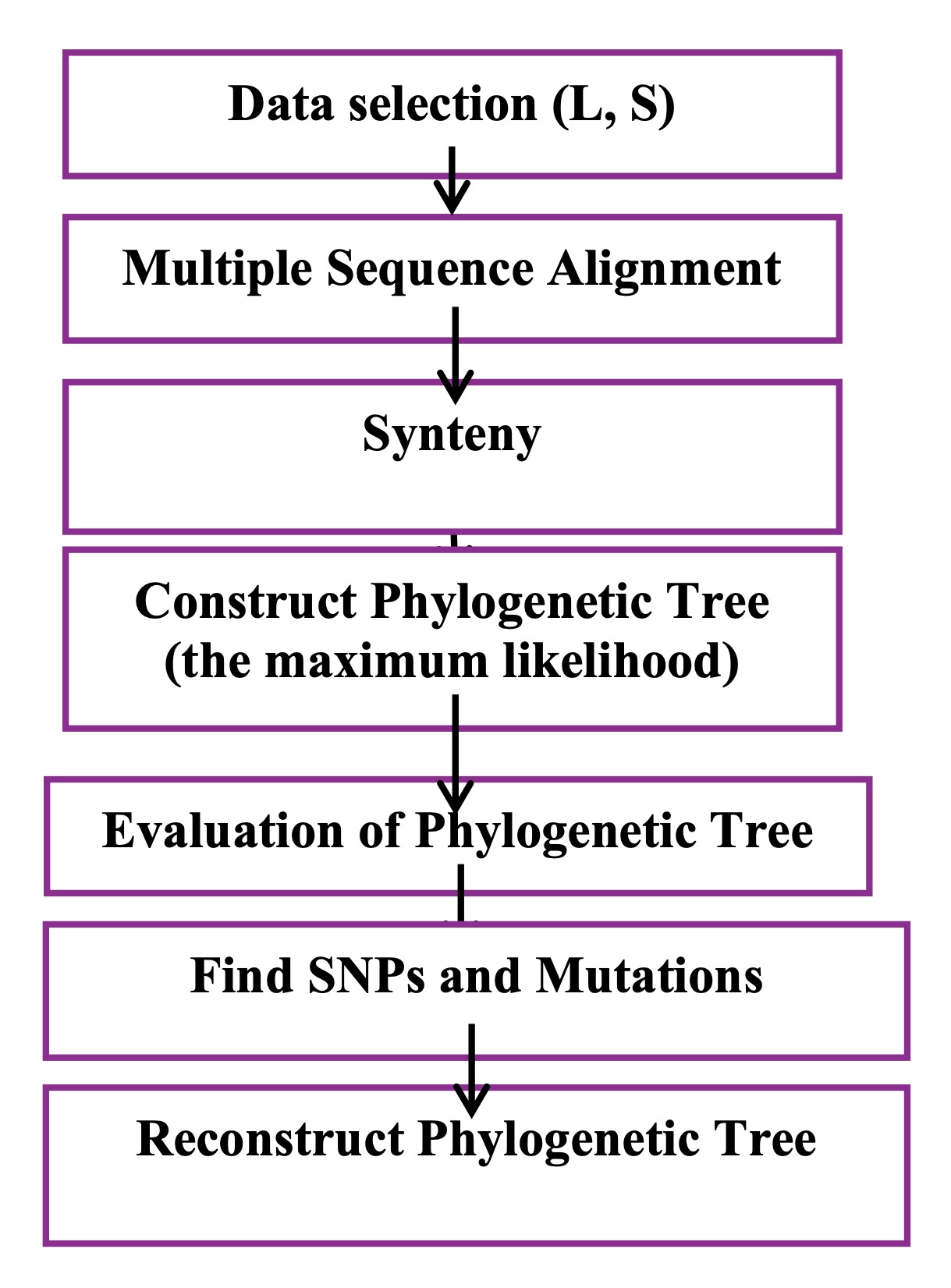
Fig. 1: Workflow of the bioinformatics analysis. Overview of the computational steps used to analyze Lassa virus genomes, from data selection and alignment to phylogenetic reconstruction and SNP/mutation analysis.
Data Selection
The data used in this paper, which are specific to the S and L segments of Lassa virus
genomes, were obtained,
selected, and refined from the National Center for Biotechnology Information (NCBI) website
[12]. These genomes
are considered complete genomes, not fragments of genomes. After the selection process was
completed, the
genomes were processed to eliminate duplicates and genomes containing ambiguous characters
(usually designated
as "N"), which could be more than 5%. At the end of this stage, we had two sets of
complete genomes
for the S and L segments. The next stage, considered an important and necessary step for
constructing reliable,
rooted genome trees, involved selecting a genome from outside the Arenaviridae family but
evolutionarily close
to it. This genome is called an out-group. In this work, an out-group genome was selected
for each segment (with
the S segment, JO4324.1 was selected, and with the L segment, the out-group NC_004291.1 was
selected).
Results and Discussion
Multiple Sequence Alignment
One of the most fundamental steps in our work is alignment, as it arranges the complete
genetic sequences and
adjusts them to the same length. This process is performed by adding gaps between the
sequences of the viral
family's genomes [13]. It is considered crucial and essential in bioinformatics for
analyzing genomes and
identifying specific differences. The results of the alignment process are used to construct
reliable genomic
trees.
It is implemented using a program called Muscle, which consists of several basic steps:
progressive alignment of
the draft, adjustment of the alignment, and an additional step, which is repeating the
stages to represent and
determine the best alignment to adopt [14]. In this research, a set of colors was used for
each nucleotide to
facilitate understanding and to clarify the differences between the genomes during
alignment, as shown in Fig.
2, which includes a and b and illustrates part of the sequence arrangement for the S and L
segment genomes.

Fig. 2: Multiple sequence alignment of Lassa virus genomes. Aligned sequences of the (a) S and (b) L segments reveal conserved regions and mutational hotspots. These differences form the basis for identifying variants that may contribute to neurotropism.
Synteny
One of the important tools that helps us understand and identify important information about
the structure of
the genome is synteny. It is a big part of genomic research [15]. This step comes after
aligning several genomes
and involves finding conserved gene configurations, looking into genome duplications, and
studying chromosomal
rearrangements. When choosing genomes and doing synteny alignment, the presence of only a
few common areas shows
that the genomes are very different, so they should not be included [16, 17]. Look at Fig. 3
(a, b), which
displays the synteny of several sequences from the Lassa S and L segment genomes by counting
how many genes are
kept in the same order. Blue indicates the least similarity, and red indicates the most
similarity. In Fig. 4
(a, b), these numbers indicate the percentage of similarity between the genomes when
compared.
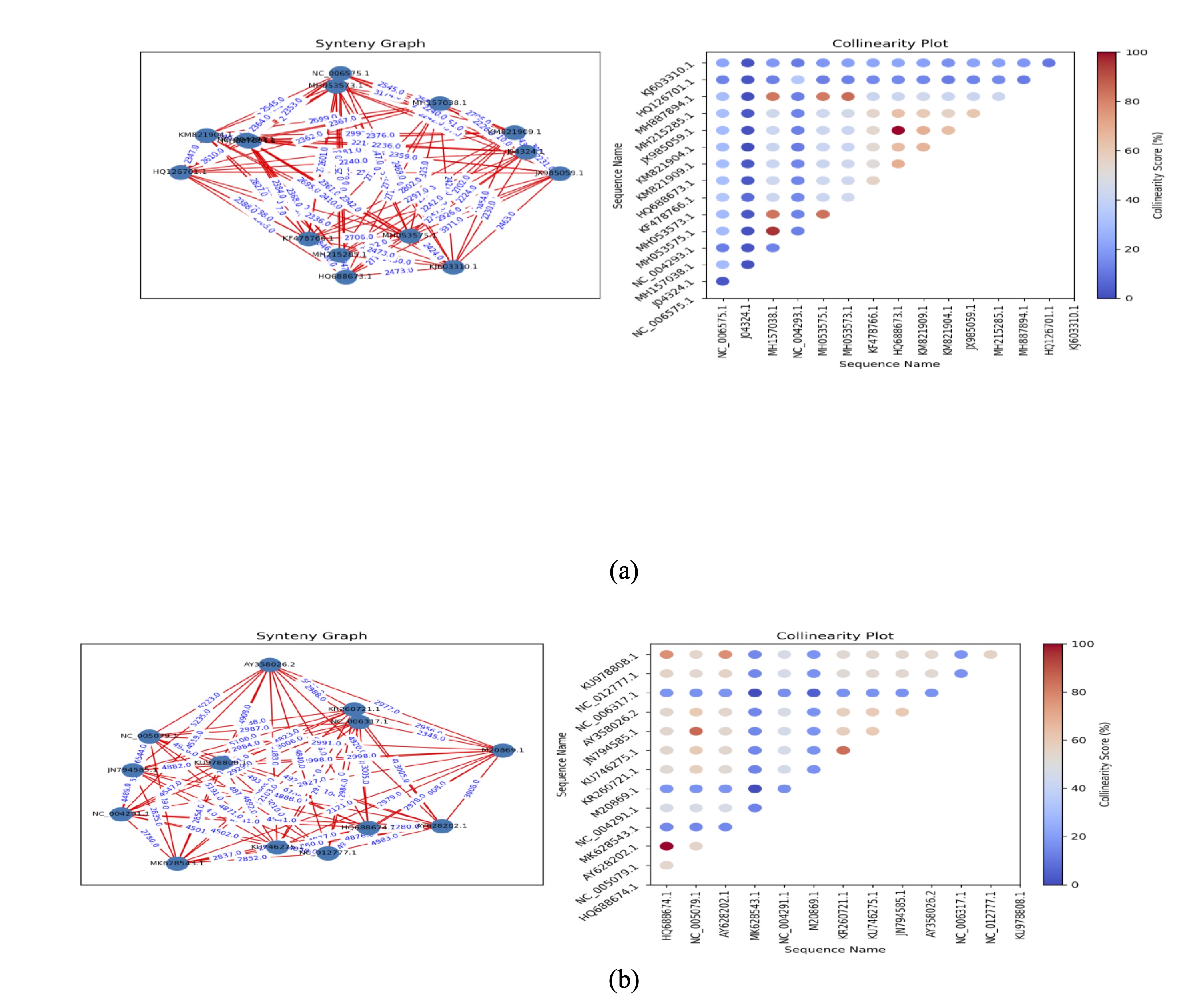
Fig. 3: Genome synteny of Lassa virus sequences. Synteny plots for the (a) S and (b) L segments highlight conserved and divergent regions across genomes, suggesting possible adaptations relevant to host interactions.
Fig. 4: Synteny heatmaps of Lassa virus genomes. Heatmaps for the (a) S and (b) L segments show degrees of similarity across viral strains. Regions of low similarity may indicate mutational hotspots linked to altered virulence.
Building Phylogenetic Trees
Molecular phylogeny is employed to examine the links among a collection of entities by
constructing a phylogenetic or evolutionary tree. The
history of evolution found in genomes shows patterns like a tree when the right data, models
for changes, and
methods for building the tree are used. These evolutionary patterns are employed to examine
the connections
among the entities [18, 19].
Before constructing the trees, a model appropriate for the genomic data was selected. This
model was GTR+G. The
genomic trees were then constructed using the maximum likelihood algorithm for both
segments, which is an
advanced statistical method. This tree is used to visualise and demonstrate the
relationships between the
genomes used. The primary benefit of these trees is to understand and illustrate how genomes
or species diverged
from a common ancestor over time. The method is powerful because it uses all available
genomes and an
appropriate evolutionary model, but it requires significant computing power [20].
Maximum Likelihood algorithm
Understanding evolutionary connections among species is essential for several biological
research projects. A
clear phylogenetic tree is crucial for understanding important changes in evolution and is
necessary for
figuring out where new genes come from, spotting molecular changes, explaining how physical
traits have evolved,
and reconstructing population changes in species that have recently split apart [21, 22].
Despite the increasing
abundance of data and the availability of robust analytical methodologies, several problems
persist in achieving
trustworthy tree construction [23].
This algorithm builds the base tree using a powerful and important program called RAxML,
along with a search
that includes the best tree [24]. Lassa virus genome segments S and L were reconstructed by
the Maximum
Likelihood method implemented in RAxML v8.2.12 [25]. Viral genomes were presented in FASTA
format, and
preliminary data processing, including sequence parsing, quality filtering, and formatting,
was performed using
Python, extensively utilising the Biopython package for modules to handle input/output on
sequences and
alignments: Bio.SeqIO and Bio.AlignIO.
We used the standalone MUSCLE tool within Python to perform multiple sequence alignment
(MSA), allowing us to
repeat and automate the procedure. The genome sequences were produced in PHYLIP format so
that RAxML could read
them. Phylogenetic trees were estimated with the GTR+G (General Time Reversible with Gamma
distribution)
substitution model, which can handle rate variation across nucleotide sites and is a better
representation of
evolutionary processes. We carried out a bootstrap analysis with 1000 repeats to evaluate
the statistical
support of the topology of the resulting tree, as bootstrap analyses are commonly used to
estimate the
robustness of inferred clades.
The most crucial command executed by RAxML was: (raxmlHPC s aligned.phy -n
output_tree -m GTRGAMMA -p
12345 -x 12345 -# 1000 -f a),Where s is the aligned input file in PHYLIP format, n gives the
name for the output
file, m GTRGAMMA chooses the GTR substitution model with gamma-distributed rate variation, p
and -x assign
random seeds for bootstrapping and tree construction, 1000 indicates the number of bootstrap
replicates, and (f,
a) allows a rapid bootstrap analysis with the discovery of the most optimal maximum
likelihood tree. After tree
reconstruction, representative final phylogenetic trees were displayed using FigTree v1.4.4.
Clades
were colored or named according to their geographic or historical aspects, and
branches were indicated
by bootstrap support values. This helped people better understand evolution.
A complex biological signal can be obtained from the tree of phylogeny produced in
this investigation,
which illustrates the evolutionary links between different Lassa virus strains. This signal
offers a rich
dataset for computational modeling, as it is expressed using branching patterns and
bootstrap support values.
These properties may be used to train algorithms that categorize viral variations, forecast
future evolutionary
trends, or discover patterns of mutational hotspots when included in artificial neural
networks. Thus, a unique
method for understanding viral evolution from a changing, data-driven point of view is
provided by coupling
phylogenetic studies with neural signal processing.
See Fig. 5, which shows the tree construction for different genomes with suitable
out-groups.
Fig. 5: Phylogenetic relationships of Lassa virus genomes. Maximum-likelihood trees of the (a) S and (b) L segments demonstrate evolutionary diversification, with several lineages associated with neurological complications.
Evaluation of Phylogenetic Tree
The tree reliability assessment process is a crucial step in determining the validity of the
selected data and
genomes, with 1000 replicates performed [26, 27]. See Fig. 6, which explains the evaluation
of the phylogenetic
tree (a) Segment S, and (b) Segment L.
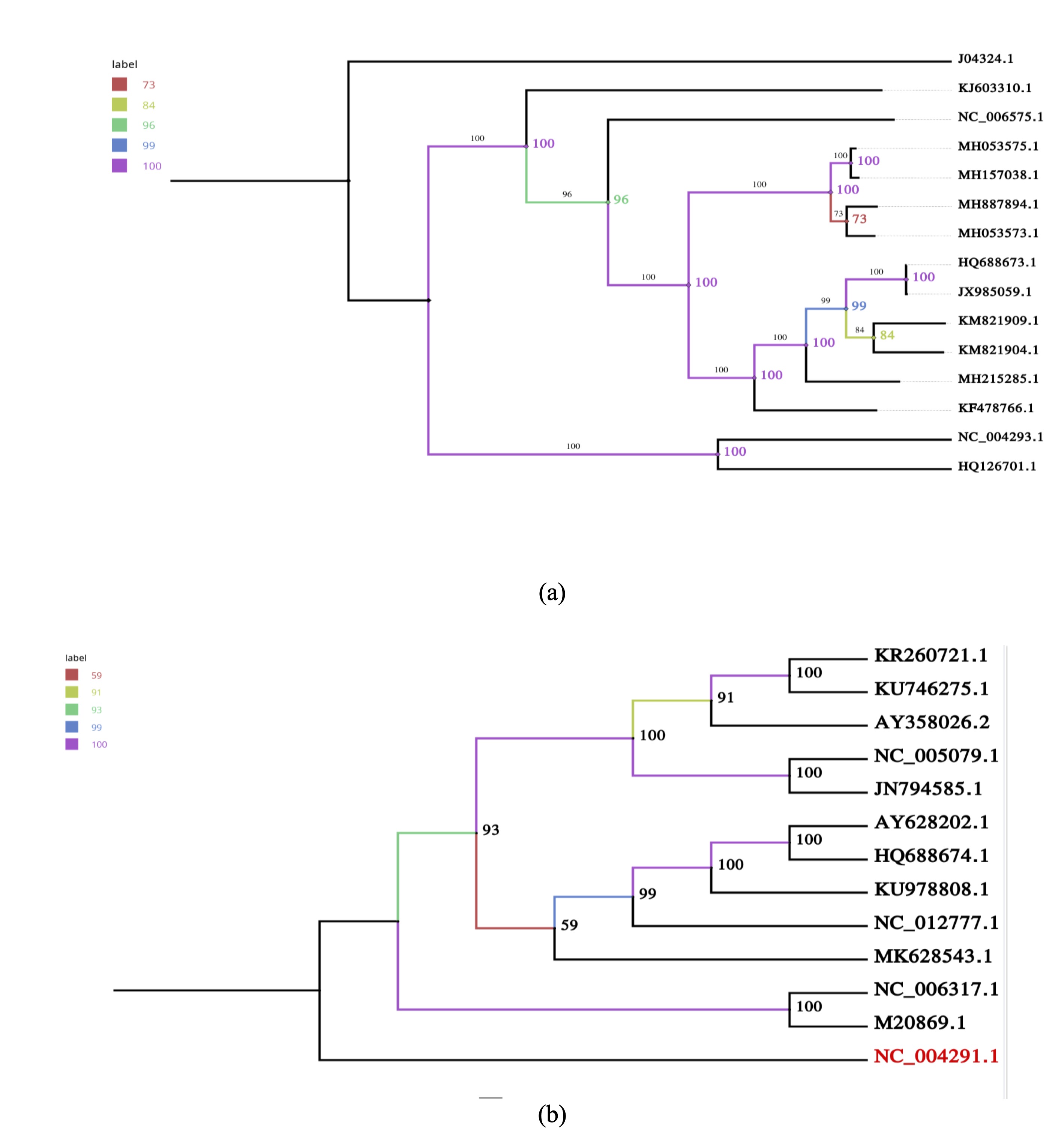
Fig. 6: Reliability of phylogenetic trees. Bootstrap analyses for the (a) S and (b) L segment trees demonstrate robustness of major clades, supporting evolutionary interpretations related to CNS involvement.
Finding SNPs and Mutations
These SNPs, their locations, and types in the genome were identified, along with their
associated mutations.
This aim was achieved by using sequence alignment to compare all virus sequences and
identify the locations of
nucleotide variations in each of them [28, 29]. Comparison was used to analyze and precisely
identify the
locations of these mutations in each gene for each virus in the analysis. Refer to Tables 1
and 2 for the
classification and quantity of SNPs and mutations included in the genomes utilized in this
study.
RAxML-NG, a more recent implementation of RAxML, was used to rebuild the ancestral genomes.
The procedure was
also enhanced with a maximum likelihood tree to perform ancestral reconstruction of genome
sequences by
comparing these genomes and identifying the most ancient common ancestor as the root of the
tree [30]. Refer to
Fig. 7, illustrating the restored ancestral tree where nodes 13 and 11 display the most
ancestral common
ancestor of the genomes used.
Each node in the phylogenetic tree generated by the study may represent an alternative
ancestral genotype,
indicating the evolutionary connections among viral genomes. To be able to train intelligent
systems like
artificial neural networks (ANNs) on phylogenetic and genomic features to forecast future
evolutionary events,
track viral diversification, and discover early warning signs of possibly harmful mutations,
the tree structure
can be viewed as a neural network, with each internal node performing as a "processing
unit" of
biological data [31]. Reconstruction of the genome sequence of the earliest known common
ancestor of Lassa virus
(both S and L segments) is an important landmark in the understanding of the evolutionary
trends of the virus.
These reference sequences provide a stable basis against which to measure genetic variation
in modern strains
and compare emerging mutations across time and space. Based on the outcome of ancestral
reconstruction and
inference of SNPs, one can frame smart predictive models using artificial intelligence and
machine learning
algorithms. These models can infer evolutionary trends and predict the rise of new mutations
or the onset of
some strains spreading to new locations [32].
The use of reconstructed ancient ancestral sequences is the foundation for building
intelligent surveillance
systems that can analyze current genetic isolates and compare them to the reference
ancestor, the root. This
enables us to detect recently occurring, potentially dangerous mutations and provide early
warning of
potentially dangerous mutations. Ultimately, this research is an effective contribution to
enabling future tools
to monitor Lassa virus with greater precision. It outlines an important framework for
improving epidemic
response and utilizing bioinformatics tools and intelligent systems to predict future viral
changes due to
various factors.
The nucleotide sequences of the genome of the oldest common ancestor in this group and of
segments S and L,
represented by nodes 13 and 11, are shown in Supplemental Tables 1 and 2. This process is
crucial for
understanding the origin of the virus and how it has evolved. This process is achieved by
observing current
genomes and how they evolved from an older common ancestor and by comparing mutations and
changes that have
occurred over time to determine which parts have persisted from the ancient ancestor to the
current genomes and
which parts have been altered as a result of various factors, possibly climatic or
environmental.
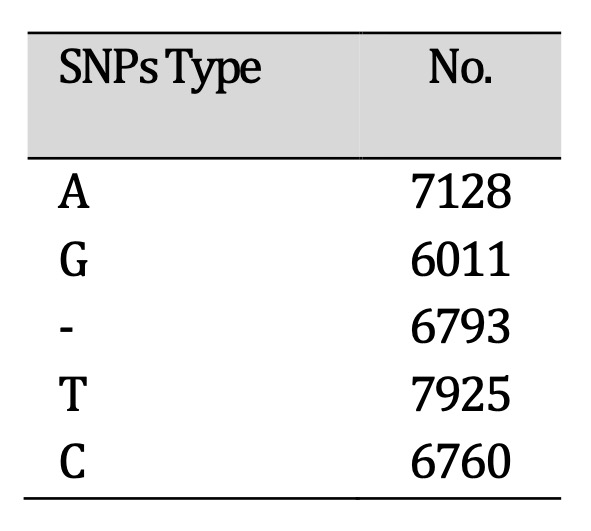
Table 1: Single nucleotide polymorphisms (SNPs) identified in Lassa virus genomes. The distribution of SNP types highlights substitution patterns that may influence viral replication and CNS involvement
Table 2: Mutational burden of Lassa virus segments. The L segment shows a particularly high mutation load, especially in the polymerase gene, which may affect replication fidelity and facilitate CNS persistence. Ancestral Sequence Reconstruction
Fig. 7: Ancestral genome reconstruction. Reconstructed sequences of (a) the S and (b) the L segments identify putative root genomes, providing references to trace mutations potentially linked to neurotropic adaptations.
The Impact of Lassa Virus Genetic Variability on Neurotropism and CNS Pathogenesis
This study reveals novel genetic variants that may influence the functioning of the brain
and spinal cord
(central nervous system). Our findings offer substantial evidence for a correlation between
the identified
mutations and neurological manifestations, despite the processes involved remaining
inadequately elucidated. The
observation that numerous identified alterations occur in genes implicated in neuroimmune
regulation, synaptic
signaling, or neuronal development supports the hypothesis that these modifications may
enhance the
vulnerability of the central nervous system to injury [33].
The neurological symptoms associated with mutations in related pathways align with the
clinical phenotypes
observed in affected patients, characterized by cognitive deficits and motor abnormalities
[34, 35]. While
experimental validation is limited, literature evidence points to similar mutations lead to
the disruption of
neuronal homeostasis, modified synaptic plasticity, or atypical neuroinflammatory responses.
These associations
underscore the need to consider CNS involvement when evaluating patients carrying these
variants [35, 36].
These mutations can cause systemic problems that make CNS disease worse since central and
peripheral symptoms
can happen at the same time. According to this perspective, mutations exert effects that are
both
cell-autonomous within neurons and non-cell-autonomous via peripheral systems, aligning with
contemporary models
of neurogenetic disorders [37].
Based on our findings, functional studies were recommended in neuronal models, and in
vivo systems must
be incorporated into future research to elucidate unique neuropathogenic pathways.
Establishing the significance
of these variations in central nervous system dysfunction and identifying potential
therapeutic targets
necessitates the integration of clinical, genetic, and mechanistic data. Overall, this study
contributes to a
growing recognition of the neurological dimension of these genetic alterations and provides
a foundation for
further neuroscience-focused investigations.
This work presents a comprehensive bioinformatics analysis of the Arenaviridae family,
focusing specifically on
the neurotropic Lassa virus (LASV) strains. Our research has identified multiple anomalies
in the viral genome
that may account for the virus's ability to infect and persist in the central nervous
system, resulting in
diverse neurological manifestations.
Genetic Variants and CNS Involvement
The identified mutations predominantly affect regions of the LASV genome that are important
for viral
replication and host cell interaction. These alterations may enhance the virus's capacity to
cross the
blood-brain barrier (BBB) or evade immune surveillance within the CNS. Similar mechanisms
have been observed in
other neurotropic viruses, where specific genetic changes facilitate neuronal invasion and
persistence. For
instance, mutations in the LASV glycoprotein precursor (GPC) can influence the virus's
ability to infect
dendritic cells, which are essential for initiating immunological responses in the central
nervous system (CNS)
[38, 39]. Additionally, the neurotropic LCMV Clone 13 strain possesses mutations in the GPC
and polymerase genes
that enhance replication within dendritic cells. This, in turn, alters immunological
responses and may affect
the central nervous system (CNS) [40, 41].
Neurological Manifestations in Lassa Fever
Common neurological symptoms in people with LASV infection include loss of sensorineural
hearing, tremors,
encephalitis, and ataxia. These symptoms may manifest during the acute period or as
post-infectious
consequences. The etiology of these symptoms is influenced by various mechanisms, including
immune-mediated
damage, metabolic disturbances, and direct viral cytotoxicity. Recent studies indicate that
the Lassa fever
virus (LASSV) significantly contributes to sensorineural hearing loss, a common and often
irreversible outcome
of the disease. Scientists think that this condition happens when viruses harm the cochlear
structures or
auditory circuits [42]. The encephalitis and ataxia observed in individuals infected with
LASV may possibly
result from inflammatory responses and viral invasion of neuronal tissues
Potential Neurological Implications of SNP Variability
The functional implications of the identified single nucleotide polymorphisms (SNPs) in the
Lassa virus genome
may influence neurological outcomes, particularly given the frequency of T and A
substitutions. Previous
research [43, 44] indicates that viral mutations can influence neurotropism, the efficacy of
viral replication
in the CNS, and the host immune response. For example, single-nucleotide polymorphisms
(SNPs) that alter viral
proteins responsible for host-cell entry or immune evasion may induce CNS symptoms such as
encephalopathy or
neuroinflammation by modifying the virus's ability to cross the blood-brain barrier or
interact with neural
cells. Changes to viral gene expression caused by deletions or substitutions in regulatory
regions may also
affect neurovirulence. These results underscore the necessity of performing targeted
functional studies to
clarify the correlation between neurological sequelae in Lassa virus infection and specific
nucleotide
modifications.
Implications of SNPs and Mutations on Neurological Outcomes
Table 2 shows that the S and L segments of the Lassa virus have a lot of mutations and
single nucleotide
polymorphisms (SNPs). The L segment shows particularly extensive mutational burden. High
mutation rates,
especially in the L segment encoding the viral RNA-dependent RNA polymerase, may influence
viral replication
fidelity, host-cell interactions, and immune evasion—factors that are increasingly
recognized as relevant to CNS
involvement [43, 44]. Mutations in regulatory or structural genes may lead to an increase in
neurotropism or
alterations in the functions of viral proteins that interact with neural cells. For
instance, alterations to the
S segment, which codes for the nucleoprotein and glycoprotein precursor, may affect viral
entry into glial cells
or neurons and how the immune system reacts in the central nervous system. Such mutational
patterns may help
explain the neurological manifestations observed in some Lassa virus infections, including
encephalopathy and
cognitive deficits.
Phylogenetic Analysis, SNPs, and Neurological Implications
This study's phylogenetic analysis provides a detailed view of the evolutionary
relationships among Lassa virus
genomes. Importantly, these ancestral reconstructions, particularly of the earliest common
ancestor of both S
and L segments, offer a reference framework to compare contemporary mutations and track
viral evolution. High
mutational burdens observed in the L segment (Table 2), combined with the diverse SNP types
identified (Table
1), suggest ongoing viral adaptation that may influence host-pathogen interactions,
including in the central
nervous system (CNS). Several changes in structural and polymerase genes may influence viral
neurotropism, the
efficacy of viral replication in neural tissue, and immune responses inside the central
nervous system (CNS).
For instance, alterations in the S segment—which encodes glycoproteins involved in host cell
entry—could
theoretically facilitate viral penetration into neuronal or glial cells, contributing to the
neurological
manifestations documented in Lassa virus infection, such as encephalopathy and cognitive
impairments [45].
The integration of phylogenetic insights with computational biology and statistical models
approaches holds
promise for predictive surveillance. Conceptualizing the phylogenetic tree as a network of
“processing units”
allows training of intelligent systems, such as artificial neural networks (ANNs), to
forecast future
evolutionary trends, identify emergent mutations, and potentially anticipate shifts that
could increase CNS
involvement. By comparing modern isolates to reconstructed ancestral sequences, AI-driven
models can detect
recently emerged, potentially neurovirulent mutations, offering an early-warning framework
for public health
interventions [31, 32]. Overall, the evolutionary history of the Lassa virus may be
elucidated, and potential
neurological risks can be anticipated, facilitated by the amalgamation of SNP analysis,
mutational mapping, and
ancestral genome reconstruction. This study integrates multiple disciplines, highlighting
the significance of
viral genomes in understanding the mechanisms by which viruses create neurological illnesses
and the potential
of contemporary bioinformatics techniques in mitigating or alleviating the severity of
virus-induced
neurological complications.
Chika-Igwenyi et al. (2021) found that Lassa fever outbreaks in Ebonyi State, Nigeria,
exhibited notable
differences in epidemiology, clinical features, and outcomes. In the beginning of the
pandemic, neurological
symptoms were rare. However, as the outbreak went on, they grew more common and were linked
to a higher death
rate and worse cases. This was notably true during the second outbreak, when a greater case
fatality rate was
also seen. This meant that the involvement of lethal strain of the virus. Our study aligns
with these findings,
as the observed neurological manifestations may stem from a molecular basis in the virus's
evolution towards
heightened neurotropism and severity, elucidated by the identified SNPs and mutations in
both the S and L
segments, alongside the reconstructed ancestral sequences [35].
McEntire et al. (2021) illustrated that a wide range of epidemic and pandemic diseases can
present with diverse
neurological manifestations, including central nervous system conditions such as meningitis,
encephalitis,
intraparenchymal hemorrhage, and seizures; peripheral and cranial nerve syndromes like
sensory neuropathy,
sensorineural hearing loss, and ophthalmoplegia; post-infectious syndromes including acute
inflammatory
polyneuropathy; and congenital syndromes such as fetal microcephaly. While some of these
diseases have
established therapies, others are managed primarily with supportive care. This perspective
complements our study
by highlighting the potential neurological implications of viral mutations, including those
identified in Lassa
virus, suggesting that specific SNPs or evolutionary changes may underline the neurological
outcomes observed in
severe cases [46].
Okokhere et al. (2016) stated that Lassa virus can cause aseptic meningitis even if there is
no bleeding.
Patients who received ribavirin exhibited favorable outcomes and did not encounter any
prolonged neurological
complications. This aligns with our findings on the central nervous system's role in Lassa
virus infections,
underscoring the necessity for prompt diagnosis and customized antiviral treatment to
prevent neurological
sequelae [47].
Saka et al. (2025) illustrated that people who survive Lassa fever often have hearing loss,
cognitive
impairment, seizures, delayed-onset paraparesis, and other neurological and sensory
problems, as well as eye and
mental problems. This study demonstrates that Lassa virus infection constitutes a
significant, enduring issue
for comprehensive treatment and rehabilitation. Our research also discovered that acute
infection might affect
the central nervous system and lead to neurological symptoms; this indicates that prompt
detection and treatment
may assist survivors in preventing long-term complications [48].
Duvignaud et al. (2020) revealed a link between Lassa fever and delayed onset paraparesis,
indicating a
potential relationship between viral infection and spinal cord injury. Patients with Lassa
fever require
meticulous neurological surveillance, as this case illustrates the extensive array of
neurological complications
that may arise weeks subsequent to acute illness. Our study indicates that the central
nervous system is engaged
during acute infection, suggesting that both acute and delayed neurological symptoms must be
considered when
assessing the disease's severity and deciding patient treatment [49].
Our study supports the notion that specific viral strains or mutations identified through
SNP and phylogenetic
analyses may contribute to the neurological manifestations of Lassa fever. This is
corroborated by Günther et
al. (2001), who detected viral RNA in cerebrospinal fluid but not in serum, suggesting that
the virus may
persist in the central nervous system and potentially influence neuropathogenesis [50].
As of now, there are no vaccines or medicines that have been licensed to stop or treat Lassa
virus infection.
However, Raabe et al. (2022) demonstrated that many vaccine platforms are in pre-clinical
development and that
many antiviral candidates show promise as treatments or post-exposure prophylactics. The
review by Raabe et al.
(2022) emphasizes clinical strategies, including exploratory treatments and hospital
engineering controls, as
pragmatic approaches to managing suspected infections. This is relevant to our study, as
understanding the
expanding therapeutic landscape and potential therapies may influence strategies for
controlling the
neurological repercussions of Lassa fever, particularly in regions experiencing current
outbreaks and among
high-risk populations [51].
Murphy and Ly (2021) emphasize that there are no vaccines or therapies that work completely
for the Lassa virus
(LASV) right now. About 37.7 million individuals in Africa are in danger of getting the
virus. In regions with
limited resources, accurate diagnosis of LASV might be difficult because the virus has a lot
of different
genetic variations and its symptoms are similar to those of other febrile infections.
Current diagnostics are
mostly laboratory-developed and not widely validated for clinical use, highlighting the
urgent need for simple,
affordable, and sensitive tests capable of distinguishing LASV lineages. Ribavirin and
supportive care are the
only drugs that have been approved for usage so far. However, ribavirin is contraindicated
during pregnancy, and
it only works in early administration. Several therapeutics and vaccines are in preclinical
development, though
very few have reached clinical testing. Continued research into LASV biology, immune
evasion, pathogenicity, and
vector ecology is crucial to guide the development of diagnostics, therapeutics, and
preventive strategies. In
light of this context, our study's focus on genetic variations and mutations is particularly
important, as it
may inform future surveillance, clinical management, and the formulation of effective
therapies [52].
Electroencephalography (EEG) has demonstrated significant potential in identifying central
nervous system (CNS)
involvement in many viral infections, including Lassa fever, encephalitis, diminished
consciousness, and seizure
management. Mueller et al. (2024) showed that EEG may effectively identify neurological
issues in high-risk,
resource-constrained environments, despite challenges such as technical artifacts,
environmental influences, and
biosafety limitations, when conducted by proficient neurophysiologists. These results
corroborate our study's
findings on central nervous system involvement linked to viral genetic variants [53],
underscoring the necessity
of using neurodiagnostic tools in the assessment of patients with Lassa virus infection.
Implications for Future Research
Experimental validation is essential to confirm the associations identified by our
bioinformatics method about
neurotropism. To elucidate how these modifications facilitate CNS invasion and persistence,
it is imperative to
do functional studies utilizing animal models, neuronal cell cultures, and advanced imaging
technologies.
Moreover, understanding the host factors that interact with these viral mutations could
provide insights into
the variability of neurological outcomes observed in LASV infections. Identifying genetic
predispositions or
immune responses that influence CNS involvement may lead to personalized therapeutic
strategies aimed at
mitigating neurological complications.
In general, the results suggest that Lassa virus (LASV) can infect the central nervous
system (CNS) because of
several mutations and genetic variations observed in the S and L segments. Our research
suggests that these
viral changes could affect neuroinvasion, persistence, and how the virus interacts with the
host's immune
system. This may elucidate the occurrence of symptoms such as encephalopathy, cognitive
deficits, and
sensorineural hearing loss after acute infections. In line with clinical observations from
earlier outbreaks and
case reports, the combination of SNP analysis, mutational mapping, and phylogenetic
reconstruction creates a
framework for following the evolution of viruses and predicting neurovirulent strains. These
results underscore
the importance of early diagnosis of CNS involvement and targeted treatment strategies,
while more experimental
validation is necessary. In summary, the findings of this study establish a foundation for
further research into
the neuropathogenesis of Lassa fever virus (LASSV) that combines bioinformatics, clinical,
and mechanistic
approaches; such research should facilitate the development of diagnostic tools, therapies,
and preventive
strategies for neurological complications associated with Lassa fever.
Conclusion
By choosing the right genomes and out-group elements for both groups, we were able to create accurate and connected trees to trace the evolutionary history of the Arenavirus family. By identifying SNPs and mutations that occurred in the origin of the virus, that is, from the ancient ancestor to the current strains—we can link these mutations to specific traits, such as drug resistance, increased disease severity, or changes in transmission. By sequencing the ancient common ancestor and identifying target regions shared by all genome lineages, we can develop treatments and vaccines. These sites could be suitable targets for vaccines or therapeutics against most genomes. We succeeded in determining how this important group of viruses is related using careful alignment, selection of appropriate models, efficient tree construction methods, and statistical analysis. Reconstructing the original genome sequences and discovering the mutations that cause these changes helps us understand how the virus has adapted and evolved, opening up new research avenues to combat the diseases it causes.
Disclosure Statement
The authors have nothing to disclose.
References
| 1 | Wratten, L., Wilm, A., & Göke, J. (2021). Reproducible, scalable, and shareable
analysis pipelines with bioinformatics workflow managers. Nature methods,
18(10), 1161-1168.
https://doi.org/10.1038/s41592-021-01254-9 |
| 2 | Radoshitzky, S. R., Buchmeier, M. J., Charrel, R. N., Gonzalez, J. P. J.,
Günther, S., Hepojoki, J., ... & Torre, J. C. D. L. (2023). ICTV virus taxonomy
profile: Arenaviridae 2023. Journal of General Virology, 104(9), 001891.
https://doi.org/10.1099/jgv.0.001891 |
| 3 | Johnson, D. M., Jokinen, J. D., Wang, M., Pfeffer, T., Tretyakova, I., Carrion
Jr, R., ... & Lukashevich, I. S. (2020). Bivalent Junin & Machupo experimental
vaccine based on alphavirus RNA replicon vector. Vaccine, 38(14), 2949-2959.
https://doi.org/10.1016/j.vaccine.2020.02.053 |
| 4 | Radoshitzky, S. R., Buchmeier, M. J., Charrel, R. N., Clegg, J. C. S., Gonzalez,
J. J., Günther, S., ... & Kuhn, J. H. (2019). ICTV virus taxonomy profile:
Arenaviridae. Journal of General Virology, 100(9),
1200-1201.https://www.microbiologyresearch.org/content/journal/jgv/10.1099/jgv.0.001280
https://doi.org/10.1099/jgv.0.001280 |
| 5 | Ehichioya, D. U., Dellicour, S., Pahlmann, M., Rieger, T., Oestereich, L.,
Becker-Ziaja, B., ... & Günther, S. (2019). Phylogeography of Lassa virus in
Nigeria. Journal of Virology, 93(21), 10-1128.
https://doi.org/10.1128/JVI.00929-19 |
| 6 | Arruda, L. B., Free, H. B., Simons, D., Ansumana, R., Elton, L., Haider, N., ...
& Kock, R. (2023). Current sampling and sequencing biases of Lassa
mammarenavirus limit inference from phylogeography and molecular epidemiology in
Lassa fever endemic regions. PLOS global public health, 3(11), e0002159.
https://doi.org/10.1371/journal.pgph.0002159 |
| 7 | Klitting, R., Mehta, S. B., Oguzie, J. U., Oluniyi, P. E., Pauthner, M. G.,
Siddle, K. J., ... & Sabeti, P. C. (2020). Lassa virus genetics. In Lassa Fever:
Epidemiology, Immunology, Diagnostics, and Therapeutics (pp. 23-65). Cham:
Springer International Publishing.
https://doi.org/10.1007/82_2020_212 |
| 8 | Klitting, R., Kafetzopoulou, L. E., Thiery, W., Dudas, G., Gryseels, S.,
Kotamarthi, A., ... & Dellicour, S. (2022). Predicting the evolution of the
Lassa virus endemic area and population at risk over the next decades. Nature
communications, 13(1), 5596.
https://doi.org/10.1038/s41467-022-33112-3 |
| 9 | Wang, X., Ye, X., Li, R., Zai, X., Hu, M., Wang, S., ... & Yue, J. (2024).
Spatio-temporal spread and evolution of Lassa virus in West Africa. BMC
infectious diseases, 24(1), 314.
https://doi.org/10.1186/s12879-024-09200-8 |
| 10 | B. Talib, H. Al-Nuaimi, and P. C. Guyeux, "Ancestral Reconstruction and
Investigations of Genomic Recombination on Chloroplast Genomes," Journal of
Integrative Bioinformatics, 2017.
|
| 11 | Solbrig, M. V. (1993). Lassa virus and central nervous system diseases. In The
Arenaviridae (pp. 325-330). Boston, MA: Springer US.
https://doi.org/10.1007/978-1-4615-3028-2_18 |
| 12 | National Centre for Biotechnology Information. (a.n.d.). National Centre for
Biotechnology Information. Retrieved July 14, 2025, from
https://www.ncbi.nlm.nih.gov/
|
| 13 | Edgar, R. C. (2021). MUSCLE v5 enables improved estimates of phylogenetic tree
confidence by ensemble bootstrapping. BioRxiv, 2021-06.
|
| 14 | Tumescheit, C., Firth, A. E., & Brown, K. (2022). CIAlign: A highly customisable
command line tool to clean, interpret and visualise multiple sequence
alignments. PeerJ, 10, e12983.
https://doi.org/10.7717/peerj.12983 |
| 15 | Liu, D., Hunt, M., & Tsai, I. J. (2018). Inferring synteny between genome
assemblies: a systematic evaluation. BMC bioinformatics, 19(1), 26.
https://doi.org/10.1186/s12859-018-2026-4 |
| 16 | Almeida-Silva, F., Zhao, T., Ullrich, K. K., Schranz, M. E., & Van de Peer, Y.
(2023). syntenet: an R/Bioconductor package for the inference and analysis of
synteny networks. Bioinformatics, 39(1), btac806.
https://doi.org/10.1093/bioinformatics/btac806 |
| 17 | Haug-Baltzell, A., Stephens, S. A., Davey, S., Scheidegger, C. E., & Lyons, E.
(2017). SynMap2 and SynMap3D: web-based whole-genome synteny browsers.
Bioinformatics, 33(14), 2197-2198.
https://doi.org/10.1093/bioinformatics/btx144 |
| 18 | Challa, S., & Neelapu, N. R. R. (2019). Phylogenetic trees: applications,
construction, and assessment. Essentials of Bioinformatics, Volume III: In
Silico Life Sciences: Agriculture, 167-192.
https://doi.org/10.1007/978-3-030-19318-8_10 |
| 19 | Kapli, P., Yang, Z., & Telford, M. J. (2020). Phylogenetic tree building in the
genomic age. Nature Reviews Genetics, 21(7), 428-444.
https://doi.org/10.1038/s41576-020-0233-0 |
| 20 | Munjal, G., Hanmandlu, M., & Srivastava, S. (2019). Phylogenetics algorithms and
applications. In Ambient Communications and Computer Systems: RACCCS-2018 (pp.
187-194). Singapore: Springer Singapore.
https://doi.org/10.1007/978-981-13-5934-7_17 |
| 21 | Mai, U., & Mirarab, S. (2018). TreeShrink: fast and accurate detection of
outlier long branches in collections of phylogenetic trees. BMC genomics,
19(Suppl 5), 272.
https://doi.org/10.1186/s12864-018-4620-2 |
| 22 | Czech, L., Huerta-Cepas, J., & Stamatakis, A. (2017). A critical review of the
use of support values in tree viewers and bioinformatics toolkits. Molecular
biology and evolution, 34(6), 1535-1542.
https://doi.org/10.1093/molbev/msx055 |
| 23 | Harmon, L. (2019). Phylogenetic comparative methods: learning from trees.
https://doi.org/10.32942/OSF.IO/E3XNR |
| 24 | Kozlov, A. M., Darriba, D., Flouri, T., Morel, B., & Stamatakis, A. (2019).
RAxML-NG: a fast, scalable and user-friendly tool for maximum likelihood
phylogenetic inference. Bioinformatics, 35(21), 4453-4455.
https://doi.org/10.1093/bioinformatics/btz305 |
| 25 | Edler, D., Klein, J., Antonelli, A., & Silvestro, D. (2021). raxmlGUI 2.0: a
graphical interface and toolkit for phylogenetic analyses using RAxML. Methods
in ecology and evolution, 12(2), 373-377.
https://doi.org/10.1111/2041-210X.13512 |
| 26 | Lemoine, F., Domelevo Entfellner, J. B., Wilkinson, E., Correia, D., Dávila
Felipe, M., De Oliveira, T., & Gascuel, O. (2018). Renewing Felsenstein's
phylogenetic bootstrap in the era of big data. Nature, 556(7702), 452-456.
https://doi.org/10.1038/s41586-018-0043-0 |
| 27 | Hoang, D. T., Chernomor, O., Von Haeseler, A., Minh, B. Q., & Vinh, L. S.
(2018). UFBoot2: improving the ultrafast bootstrap approximation. Molecular
biology and evolution, 35(2), 518-522.
https://doi.org/10.1093/molbev/msx281 |
| 28 | Leaché, A. D., & Oaks, J. R. (2017). The utility of single-nucleotide
polymorphism (SNP) data in phylogenetics. Annual review of ecology, evolution,
and systematics, 48, 69-84.
https://doi.org/10.1146/annurev-ecolsys-110316-022645 |
| 29 | Singer, J., Kuipers, J., Jahn, K., & Beerenwinkel, N. (2018). Single-cell
mutation identification via phylogenetic inference. Nature communications, 9(1),
5144.
https://doi.org/10.1038/s41467-018-07627-7 |
| 30 | Spence, M. A., Kaczmarski, J. A., Saunders, J. W., & Jackson, C. J. (2021).
Ancestral sequence reconstruction for protein engineers. Current opinion in
structural biology, 69, 131-141.
https://doi.org/10.1016/j.sbi.2021.04.001 |
| 31 | Samson, T. K., Akingbade, T., & Orija, J. (2023). Comparative analysis of
mortality predictions from Lassa fever in Nigeria: A study using count
regression and machine learning methods. Acadlore Transactions on AI and Machine
Learning, 2(4), 204-211.
https://doi.org/10.56578/ataiml020403 |
| 32 | Ishikawa, S. A., Zhukova, A., Iwasaki, W., & Gascuel, O. (2019). A fast
likelihood method to reconstruct and visualise ancestral scenarios. Molecular
biology and evolution, 36(9), 2069-2085.
https://doi.org/10.1093/molbev/msz131 |
| 33 | Wongchitrat, P., Chanmee, T. & Govitrapong, P. Molecular Mechanisms Associated
with Neurodegeneration of Neurotropic Viral Infection. Mol Neurobiol 61,
2881-2903 (2024). https://doi.org/10.1007/s12035-023-03761-6
https://doi.org/10.1007/s12035-023-03761-6 |
| 34 | Safronetz, D., Strong, J. E., Feldmann, F., Haddock, E., Sogoba, N., Brining,
D., Geisbert, T. W., Scott, D. P., & Feldmann, H. (2013). A recently isolated
Lassa virus from Mali demonstrates atypical clinical disease manifestations and
decreased virulence in cynomolgus macaques. Journal of Infectious Diseases,
207(8), 1316-1327. https://doi.org/10.1093/infdis/jit004
https://doi.org/10.1093/infdis/jit004 |
| 35 | Chika-Igwenyi, N. M., Harrison, R. E., Psarra, C., Gil-Cuesta, J., Gulamhusein,
M., Onwe, E. O., Onoh, R. C., Unigwe, U. S., Ajayi, N. A., Nnadozie, U. U.,
Ojide, C. K., Nwidi, D. U., Ezeanosike, O., Sampson, E., Adeke, A. S., Ugwu, C.
N., Anebonam, U., Tshiang, J. K., Maikere, J., & Reid, A. (2021). Early onset of
neurological features differentiates two outbreaks of Lassa fever in Ebonyi
state, Nigeria during 2017-2018. PLoS Neglected Tropical Diseases, 15(3),
e0009169. https://doi.org/10.1371/journal.pntd.0009169
https://doi.org/10.1371/journal.pntd.0009169 |
| 36 | Löscher, W., & Howe, C. L. (2022). Molecular mechanisms in the genesis of
seizures and epilepsy associated with viral infection. Frontiers in Molecular
Neuroscience, 15, 870868. https://doi.org/10.3389/fnmol.2022.870868
https://doi.org/10.3389/fnmol.2022.870868 |
| 37 | Verheijen, B. M., Vermulst, M., & van Leeuwen, F. W. (2018). Somatic mutations
in neurons during aging and neurodegeneration. Acta Neuropathologica, 135(6),
811-826. https://doi.org/10.1007/s00401-018-1850-y
https://doi.org/10.1007/s00401-018-1850-y |
| 38 | Loureiro, M. E., D'Antuono, A., & López, N. (2019). Virus-Host Interactions
Involved in Lassa Virus Entry and Genome Replication. Pathogens, 8(1), 17.
https://doi.org/10.3390/pathogens8010017
https://doi.org/10.3390/pathogens8010017 |
| 39 | Zapata, J. C., & Salvato, M. S. (2013). Arenavirus Variations Due to
Host-Specific Adaptation. Viruses, 5(1), 241-278.
https://doi.org/10.3390/v5010241
https://doi.org/10.3390/v5010241 |
| 40 | Sullivan, B. M., Emonet, S. F., Welch, M. J., Lee, A. M., Campbell, K. P., de la
Torre, J. C., & Oldstone, M. B. (2011). Point mutation in the glycoprotein of
lymphocytic choriomeningitis virus is necessary for receptor binding, dendritic
cell infection, and long-term persistence. Proceedings of the National Academy
of Sciences of the United States of America, 108(7), 2969-2974.
https://doi.org/10.1073/pnas.1019304108
https://doi.org/10.1073/pnas.1019304108 |
| 41 | Plume, J. M., Todd, D., & Bonthius, D. J. (2019). Viral strain determines
disease symptoms, pathology, and immune response in neonatal rats with
lymphocytic choriomeningitis virus infection. Viruses, 11(6), 552.
https://doi.org/10.3390/v11060552
https://doi.org/10.3390/v11060552 |
| 42 | Mateer, E. J., Huang, C., Shehu, N. Y., & Paessler, S. (2018). Lassa
fever-induced sensorineural hearing loss: A neglected public health and social
burden. PLoS Neglected Tropical Diseases, 12(2), e0006187.
https://doi.org/10.1371/journal.pntd.0006187
https://doi.org/10.1371/journal.pntd.0006187 |
| 43 | Taniguchi, S., Saito, T., Paroha, R., Huang, C., Paessler, S., & Maruyama, J.
(2024). Unraveling factors responsible for pathogenic differences in Lassa virus
strains. bioRxiv. https://doi.org/10.1101/2024.05.21.595091
https://doi.org/10.1101/2024.05.21.595091 |
| 44 | Kolawole, D., Raji, H., & Okeke, M. I. (2021). Phylogenetic and mutational
analysis of Lassa virus strains isolated in Nigeria: Proposal for an in silico
study. JMIR Research Protocols, 10(3), e23015. https://doi.org/10.2196/23015
https://doi.org/10.2196/23015 |
| 45 | Pennington, H. N., & Lee, J. (2022). Lassa virus glycoprotein complex review:
Insights into its unique fusion machinery. Bioscience Reports, 42(2),
BSR20211930. https://doi.org/10.1042/BSR20211930
https://doi.org/10.1042/BSR20211930 |
| 46 | McEntire, C. R. S., Song, K.-W., McInnis, R. P., Rhee, J. Y., Young, M.,
Williams, E., Wibecan, L. L., Nolan, N., Nagy, A. M., Gluckstein, J., Mukerji,
S. S., & Mateen, F. J. (2021). Neurologic manifestations of the World Health
Organization's list of pandemic and epidemic diseases. Frontiers in Neurology,
12, 634827. https://doi.org/10.3389/fneur.2021.634827
https://doi.org/10.3389/fneur.2021.634827 |
| 47 | Okokhere, P. O., Bankole, I. A., Iruolagbe, C. O., Muoebonam, B. E., Okonofua,
M. O., Dawodu, S. O., & Akpede, G. O. (2016). Aseptic meningitis caused by Lassa
virus: Case series report. Case Reports in Neurological Medicine, 2016, 1978461.
https://doi.org/10.1155/2016/1978461
https://doi.org/10.1155/2016/1978461 |
| 48 | Saka, S. A., Lawal, Q. O., Otaigbe, O., Blackie, F. F., Ighodaro, O., Odafen, P.
I., & Okogbenin, S. (2025). Lassa fever survivors: Long-term health effects and
chronic sequelae - a scoping review. BMC Infectious Diseases, 25(1), 823.
https://doi.org/10.1186/s12879-025-11262-1
https://doi.org/10.1186/s12879-025-11262-1 |
| 49 | Duvignaud, A., Doutchi, M., Abejegah, C., Etafo, I., Jaspard, M., Serra, B.,
Tricaud, E., Levy-Marchal, C., Anglaret, X., Ahmed, L. A., Adedosu, A. N.,
Malvy, D., & Ayodeji, O. O. (2020). Delayed-onset paraparesis in Lassa fever: A
case report. International Journal of Infectious Diseases, 92, 49-52.
https://doi.org/10.1016/j.ijid.2019.12.022
https://doi.org/10.1016/j.ijid.2019.12.022 |
| 50 | Günther, S., Weisner, B., Roth, A., Grewing, T., Asper, M., Drosten, C.,
Emmerich, P., Petersen, J., Wilczek, M., & Schmitz, H. (2001). Lassa fever
encephalopathy: Lassa virus in cerebrospinal fluid but not in serum. Journal of
Infectious Diseases, 184(3), 345-349. https://doi.org/10.1086/322033
https://doi.org/10.1086/322033 |
| 51 | Raabe, V., Mehta, A. K., Evans, J. D., Beitscher, A., Bhadelia, N., Brett-Major,
D., Cieslak, T. J., Davey, R. T., Evans, J. D., Frank, M. G., Iwen, P.,
Kortepeter, M. G., Levine, C., McLellan, S., Mehta, A. K., Sauer, L., Shenoy, E.
S., & Zachary, K. (2022). Lassa virus infection: A summary for clinicians.
International Journal of Infectious Diseases, 119, 187-200.
https://doi.org/10.1016/j.ijid.2022.04.004
https://doi.org/10.1016/j.ijid.2022.04.004 |
| 52 | Murphy, H. L., & Ly, H. (2021). Pathogenicity and virulence mechanisms of Lassa
virus and its animal modeling, diagnostic, prophylactic, and therapeutic
developments. Virulence, 12(1), 2989-3014.
https://doi.org/10.1080/21505594.2021.2000290
https://doi.org/10.1080/21505594.2021.2000290 |
| 53 | Mueller, H. C. S., Erameh, C. O., Gelderblom, M., Edeawe, O. I., Akpasubi, O.
G., Ekoyata, E. U., et al. (2024). Electroencephalography in emerging viral
infections: Lessons learned from implementing an EEG unit in a Lassa fever
isolation ward in Nigeria. PLoS Neglected Tropical Diseases, 18(10), e0012522.
https://doi.org/10.1371/journal.pntd.0012522
https://doi.org/10.1371/journal.pntd.0012522 |